PTM Analysis - N-to-C Plots
dpa Analysis
FGCZ
29 July, 2026
Source:vignettes/Analysis_n_to_c.Rmd
Analysis_n_to_c.RmdIntroduction
This document generates N-to-C plots for DPA analysis.
desc <- switch(params$analysis_type,
"dpa" = "**DPA (Differential PTM Abundance)**: Raw PTM signal changes without protein abundance correction. Shows both site-level and protein-level fold changes.",
"dpu" = "**DPU (Differential PTM Usage)**: Protein-normalized PTM changes (model-first approach). Reflects genuine modification stoichiometry changes.",
"cf" = "**CF (CorrectFirst)**: Alternative protein-correction approach where abundances are normalized before statistical modeling."
)
cat(desc)DPA (Differential PTM Abundance): Raw PTM signal changes without protein abundance correction. Shows both site-level and protein-level fold changes.
Data Loading
if (pipeline_mode) {
message("Loading data from: ", params$xlsx_file, " (sheet: ", params$sheet, ")")
data <- readxl::read_xlsx(params$xlsx_file, sheet = params$sheet)
output_dir <- if (!is.null(params$output_dir)) params$output_dir else dirname(params$xlsx_file)
} else {
# Vignette mode: use example data
data("combined_test_diff_example", package = "prophosqua")
data <- combined_test_diff_example
output_dir <- tempdir()
max_fig <- 5
message("Using example data from prophosqua package")
}
data_info <- tibble(
Property = c("Mode", "Sheet", "Analysis Type", "Rows", "Contrasts"),
Value = c(
if (pipeline_mode) basename(params$xlsx_file) else "Example data",
params$sheet, toupper(params$analysis_type),
nrow(data), paste(unique(data$contrast), collapse = ", ")
)
)
knitr::kable(data_info, caption = "Data Summary")| Property | Value |
|---|---|
| Mode | Example data |
| Sheet | DPA |
| Analysis Type | DPA |
| Rows | 105824 |
| Contrasts | KO_vs_WT, KO_vs_WT_at_Early, KO_vs_WT_at_Late, KO_vs_WT_at_Uninfect |
# Prepare data for plotting using shared function
plot_data <- prepare_ntoc_data(data, params$analysis_type)
all_contrasts <- unique(plot_data$contrast)
cat("Found", length(all_contrasts), "contrasts:", paste(all_contrasts, collapse = ", "), "\n")## Found 4 contrasts: KO_vs_WT, KO_vs_WT_at_Early, KO_vs_WT_at_Late, KO_vs_WT_at_UninfectGenerate N-to-C Plots
Filtering criteria: Proteins are included if they contain at least one PTM site with FDR < 0.05 and |log2 fold change| > 0.6.
if (params$analysis_type == "dpa") {
# DPA: Use expression plotting (shows protein + site)
plot_result <- n_to_c_expression_multicontrast(
plot_data,
FDR_threshold = params$fdr,
fc_threshold = params$fc,
max_plots = max_fig
)
} else {
# DPU/CF: Use usage plotting (shows site only)
plot_result <- n_to_c_usage_multicontrast(
plot_data,
FDR_threshold = params$fdr,
fc_threshold = params$fc,
max_plots = max_fig
)
}
n_plots <- nrow(plot_result)
stopifnot("No significant sites found. Adjust fdr/log2fc thresholds in config." = n_plots > 0)
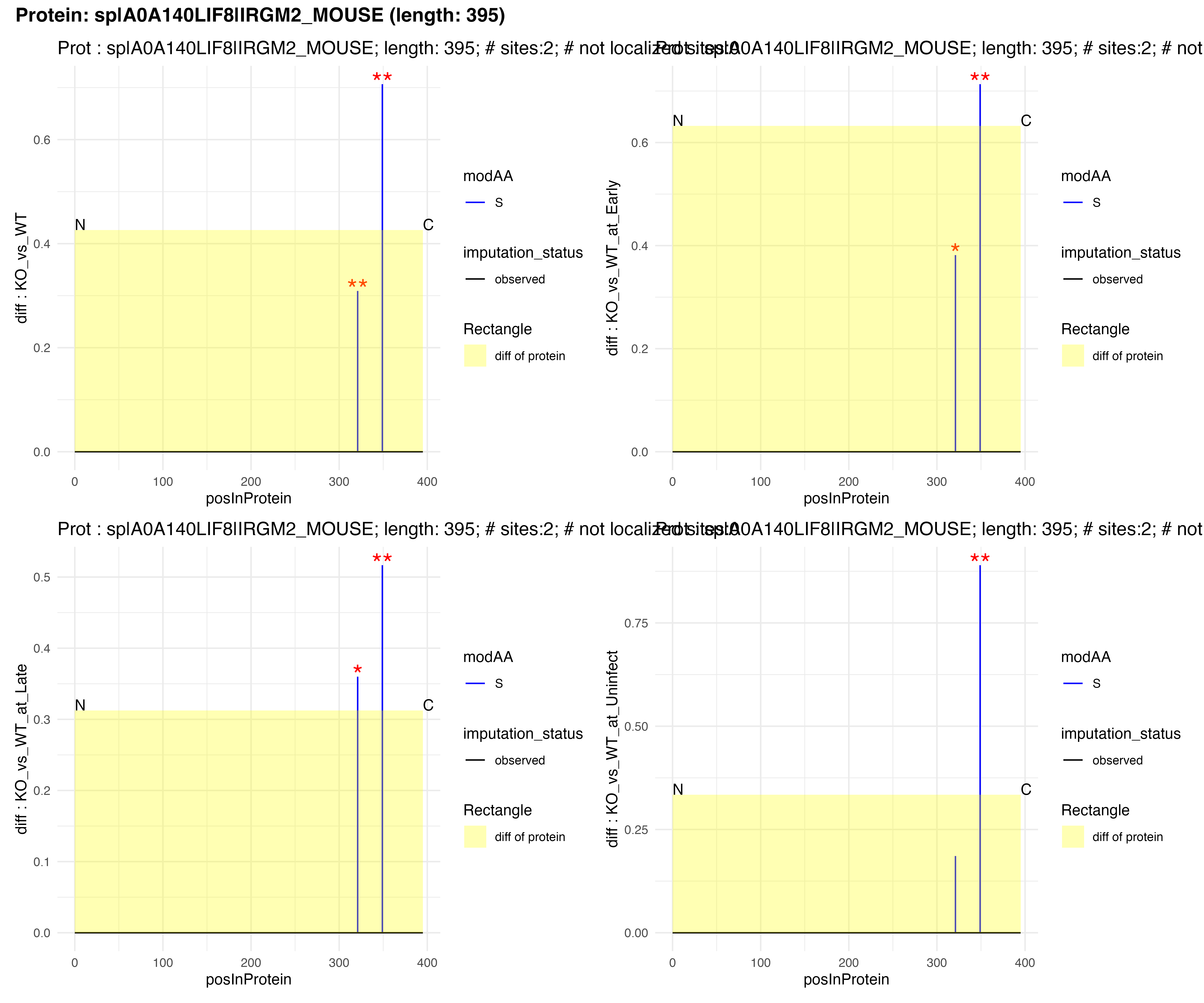
cat("Generated", n_plots, "multi-contrast plots\n")Example Plots
Showing example plots in the HTML report: one protein WITH protein-level data and one WITHOUT (if available).
if (n_plots > 0 && params$analysis_type == "dpa") {
# For DPA: show examples of proteins with and without protein-level data
# Find proteins with matched protein data (has diff.protein)
matched_proteins <- plot_data |>
dplyr::filter(!is.na(diff.protein)) |>
dplyr::pull(protein_Id) |>
unique()
# Find proteins without matched protein data
unmatched_proteins <- plot_data |>
dplyr::filter(is.na(diff.protein)) |>
dplyr::pull(protein_Id) |>
unique()
# Get indices in plot_result for each type
matched_idx <- which(plot_result$protein_Id %in% matched_proteins)
unmatched_idx <- which(plot_result$protein_Id %in% unmatched_proteins)
examples_to_show <- c()
# Add one matched protein if available
if (length(matched_idx) > 0) {
examples_to_show <- c(examples_to_show, matched_idx[1])
}
# Add one unmatched protein if available
if (length(unmatched_idx) > 0) {
examples_to_show <- c(examples_to_show, unmatched_idx[1])
}
# Fallback: if no examples found, use first 2
if (length(examples_to_show) == 0) {
examples_to_show <- seq_len(min(2, n_plots))
}
cat("\n\n**Note:** ", length(matched_idx), " proteins have protein-level data, ",
length(unmatched_idx), " proteins do not.\n\n", sep = "")
for (i in examples_to_show) {
has_protein <- plot_result$protein_Id[i] %in% matched_proteins
label <- if (has_protein) "(with protein data)" else "(NO protein data)"
cat("\n\n### ", plot_result$protein_Id[i], " ", label, "\n\n", sep = "")
print(plot_result$plot[[i]])
cat("\n\n")
}
} else if (n_plots > 0) {
# DPU/CF: just show first 2
n_examples <- min(2, n_plots)
for (i in seq_len(n_examples)) {
cat("\n\n### ", plot_result$protein_Id[i], "\n\n")
print(plot_result$plot[[i]])
cat("\n\n")
}
} else {
cat("No significant proteins found with the current filtering criteria.\n")
}Note: 5 proteins have protein-level data, 0 proteins do not.
Export Plots
# Use explicit output_dir if provided, otherwise use xlsx directory
pdf_dir <- output_dir
if (!dir.exists(pdf_dir)) {
dir.create(pdf_dir, recursive = TRUE)
}
# Determine output filename based on analysis type
pdf_name <- switch(params$analysis_type,
"dpa" = "Site_differential_Expression_MultiContrast.pdf",
"dpu" = "Site_differential_UsageChange_MultiContrast.pdf",
"cf" = "Site_differential_CorrectFirst_MultiContrast.pdf"
)
pdf_path <- file.path(pdf_dir, pdf_name)
if (n_plots > 0) {
pdf(pdf_path, width = 14, height = 10)
for (i in seq_len(n_plots)) {
print(plot_result$plot[[i]])
}
dev.off()
cat("Exported", n_plots, "plots to:", pdf_path, "\n")
} else {
cat("No plots to export.\n")
}
message("Vignette mode: PDF export skipped.")
pdf_path <- NULLResults Summary
summary_info <- tibble(
Metric = c("Analysis Type", "Total Proteins", "Plots Generated",
"Shown in HTML", "FDR Threshold", "FC Threshold"),
Value = c(toupper(params$analysis_type), n_distinct(plot_data$protein_Id),
n_plots, min(2, n_plots), params$fdr, params$fc)
)
knitr::kable(summary_info, caption = "Analysis Summary")| Metric | Value |
|---|---|
| Analysis Type | DPA |
| Total Proteins | 9581 |
| Plots Generated | 5 |
| Shown in HTML | 2 |
| FDR Threshold | 0.05 |
| FC Threshold | 0.6 |
if (pipeline_mode && n_plots > 0) {
cat("\nPDF exported to:", pdf_path, "\n")
}Session Info
## R version 4.6.1 (2026-06-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] patchwork_1.3.2 readxl_1.5.0 dplyr_1.2.1 prophosqua_0.3.0
##
## loaded via a namespace (and not attached):
## [1] gtable_0.3.6 jsonlite_2.0.0 compiler_4.6.1 tidyselect_1.2.1
## [5] jquerylib_0.1.4 systemfonts_1.3.2 scales_1.4.0 textshaping_1.0.5
## [9] yaml_2.3.12 fastmap_1.2.0 ggplot2_4.0.3 R6_2.6.1
## [13] labeling_0.4.3 generics_0.1.4 knitr_1.51 htmlwidgets_1.6.4
## [17] forcats_1.0.1 tibble_3.3.1 bookdown_0.47 desc_1.4.3
## [21] RColorBrewer_1.1-3 bslib_0.11.0 pillar_1.11.1 rlang_1.3.0
## [25] cachem_1.1.0 xfun_0.60 S7_0.2.2 fs_2.1.0
## [29] sass_0.4.10 otel_0.2.0 cli_3.6.6 withr_3.0.3
## [33] pkgdown_2.2.1 magrittr_2.0.5 digest_0.6.39 grid_4.6.1
## [37] lifecycle_1.0.5 vctrs_0.7.3 evaluate_1.0.5 glue_1.8.1
## [41] cellranger_1.1.0 farver_2.1.2 ggseqlogo_0.2.2 ragg_1.5.2
## [45] purrr_1.2.2 rmarkdown_2.31 tools_4.6.1 pkgconfig_2.0.3
## [49] htmltools_0.5.9