Integrated Analysis of Post-Translational Modifications and Total Proteome: Methods for Distinguishing Abundance from Usage Changes
Witold Wolski
Antje Dittmann
Christian Panse
Laura Kunz
Jonas Grossmann
2025-07-15
Source:vignettes/MiMBIntegratedPTM.Rmd
MiMBIntegratedPTM.Rmd| Author | Affiliation(s) | Corresponding Author Email |
|---|---|---|
| Witold Wolski | FGCZ, SIB | witold.wolski@fgcz.uzh.ch |
| Antje Dittmann | FGCZ | antje.dittmann@fgcz.uzh.ch |
| Laura Kunz | FGCZ | |
| Christian Panse | FGCZ, SIB | |
| Jonas Grossmann | FGCZ, SIB | jonas.grossmann@fgcz.uzh.ch |
- FGCZ - Functional Genomics Center Zurich, Winterthurerstrasse 190, CH-8057 Zurich,
- SIB - Swiss Institute of Bioinformatics, Winterthurerstrasse 190, CH-8057 Zurich
Abstract
Post-translational modifications (PTMs), particularly phosphorylation, regulate protein function at substoichiometric levels, making their quantitative analysis technically challenging. A critical limitation in PTM studies is distinguishing between changes in modification abundance due to altered protein expression versus genuine changes in modification stoichiometry (the fraction of protein molecules that are modified).
This chapter presents a comprehensive workflow integrating
Tandem-Mass-Tag (TMT)-based quantitative proteomics with specialized
computational analysis to distinguish differential PTM abundance (DPA)
from differential PTM usage (DPU) using the prolfqua,
prolfquapp, and prophosqua R
packages. The workflow introduces a robust statistical framework for
integrated PTM-protein analysis, comprehensive visualization tools
including N-terminus to C-terminus (N-to-C) plots for protein-centric
PTM mapping, and sequence motif analysis for kinase prediction.
This integrated approach enables researchers to identify actual signaling changes in PTM studies, prioritize functionally relevant modification sites, understand pathway-level regulation in disease contexts, and generate robust datasets suitable for publication in high-impact journals.
Key Words
Post-translational modifications, phosphoproteomics, TMT labeling, differential PTM abundance analysis, differential PTM usage analysis, mass spectrometry, data integration, kinase activity
Introduction
Biological Significance of Post-Translational Modifications
Protein phosphorylation is a reversible PTM that modulates a protein’s conformation, enzymatic activity, subcellular localization, or binding interactions. Because this modification can be reversed rapidly, it acts as a molecular switch in cellular signaling networks. As a result, cells respond to environmental cues without requiring new protein synthesis. Dysregulation of phosphorylation is implicated in cancer, neurodegeneration, and metabolic disorders [1]. Large-scale phosphoproteomic profiling by high-throughput mass spectrometry (MS) (e.g., TMT-based workflows) is therefore crucial for dissecting disease-associated signaling alterations and identifying candidate therapeutic targets.
Technical Challenges in PTM Analysis
The analysis of PTMs, such as phosphorylation, presents significant analytical challenges due to the low abundance of modified peptides. Typically, phosphorylated peptides represent less than 1–5% of the total peptide population [2]. Additionally, phosphopeptides generally exhibit poorer ionization efficiency in MS-based proteomics compared to non-modified peptides, further complicating their detection and quantification. This low abundance and challenging ionization behavior necessitate specialized experimental workflows and computational strategies to achieve accurate quantification [3].
A key analytical challenge is distinguishing genuine changes in PTM levels, due to altered kinase and phosphatase activity, from changes caused by altered protein abundance [4, 5]. Without proper correction, this confounding factor can lead researchers to misinterpret PTM data, incorrectly attributing higher phosphorylation signals solely to increased modification efficiency or pathway activation, rather than to elevated protein expression [6]. Controlling for this confounder is critical to discern rapid, signaling-driven phosphorylation events from slower, transcriptionally or translationally regulated changes in protein abundance.
Because phosphorylation is substoichiometric, only a small fraction of protein molecules carry the modification; therefore, enrichment is required to isolate phosphopeptides. Enrichment steps (such as Immobilized Metal Affinity Chromatography (IMAC) or Metal Oxide Affinity Chromatography (MOAC)) introduce additional sample handling and technical variability [6–8]. Therefore, each stage, from digestion to cleanup to MS acquisition, must be carefully optimized to improve reproducibility [9]. Effective experimental design combined with tailored normalization strategies is therefore essential to recover reliable, biologically meaningful PTM signals.
Isobaric TMT labeling enables multiplexed PTM analysis of up to 35 samples in a single run, reducing inter-sample variability and missing values to improve statistical power [10, 11]. Phosphopeptides are typically enriched by IMAC or Metal Oxide Affinity Chromatography (MOAC), which provide complementary selectivity and broaden coverage [12, 13]. Adding offline fractionation before liquid chromatography–tandem mass spectrometry (LC–MS/MS) further increases phosphosite depth and distributes peptide load, improving quantification consistency across fractions and conditions [14].
In TMT-based proteomics, a plex refers to the complete set of isobarically tagged samples analyzed together in a single LC–MS/MS run. When the total number of samples exceeds the available TMT channels, each plex should include at least one biological replicate from every experimental group and a common reference or pooled channel for normalization [15]. Replicates of the same group are then distributed across different plexes rather than clustered within a single run, preventing any group’s replicates from being confined to one batch. If all samples fit within the channel capacity of a single plex, these balancing measures are inherently satisfied in that run.
Computational Tools and Workflows
The computational landscape for quantitative protein PTM analysis has undergone significant evolution, with numerous specialized software platforms supporting bottom-up proteomics workflows. Following MS data acquisition, spectra undergo database searching to identify peptide sequences, which is subsequently followed by protein inference to determine protein identities based on these identified peptides. The ecosystem of PTM analysis includes several well-established data-dependent acquisition (DDA)-TMT compatible software suites, both free and commercial, such as Andromeda integrated within MaxQuant [16], Proteome Discoverer (Thermo Fisher Scientific), FragPipe [17], or PeptideShaker [18].
Among the analytical steps following peptide identification, PTM site localization scoring represents a particularly crucial computational challenge. Site localization determines the confidence with which a modification can be assigned to a specific amino acid residue within a peptide sequence. This process is especially challenging when peptides contain multiple potential modification sites, necessitating algorithms to evaluate the quality and specificity of site-determining fragment ions. Specialized tools have been developed explicitly for robust PTM site localization, including PTMProphet [19], PhosphoRS [20], and Ascore [21]. Accurate localization is essential, as incorrect site assignments can lead to biological misinterpretations, particularly in signaling pathway analyses and kinase-substrate predictions.
In DDA workflows, FragPipe [22] represents a widely adopted and integrated platform that comprehensively addresses PTM analysis, including site localization challenges. FragPipe integrates the MSFragger search engine [23], enabling highly sensitive peptide identification, and employs PTMProphet [24] functionality for site localization scoring. It also generates detailed quantitative outputs, including site-level quantification reports and multisite feature reports that group peptides sharing identical modification patterns. A detailed description of the FragPipe workflow is provided in the experimental methods section (see Section @ref(fragpipeparams)).
While the above discussion has focused primarily on DDA workflows, data-independent acquisition (DIA)-based approaches represent alternative strategies with distinct computational considerations for PTM analysis. Specialized DIA platforms such as Spectronaut [25], [26] and DIA-NN [27] offer PTM support through dedicated site-localization scoring algorithms and site-level quantification capabilities. Unlike DDA, DIA workflows require specialized computational approaches to extract and quantify modification-specific information from highly multiplexed fragmentation spectra. Both Spectronaut and DIA-NN incorporate advanced statistical frameworks designed explicitly for PTM identification and quantification in DIA workflows.
Data Analysis Frameworks
Proteomic data analysis can be conducted at multiple levels of granularity, each providing distinct analytical perspectives. A peptidoform represents a specific peptide sequence with a particular set of modifications, while site-level analysis aggregates signals for each residue position across multiple peptides. Differential analysis can therefore target either peptidoforms or individual modification sites, with each approach offering unique advantages for different research questions.
FragPipe introduces an additional analytical concept through multisite features, which refers to the set of all identified peptide-forms sharing the same set of modification sites under investigation, regardless of sequence derivatives, cleavage state, or whether the modifications are unambiguously localized. This approach groups peptidoforms with different sequences, which can arise from missed cleavages or alternative cleavage patterns, but identical possible modification sites into the same multisite feature, providing a balanced approach between peptidoform specificity and site-level aggregation. Discarding all peptidoforms with ambiguous localization would result in the loss of a large fraction of PTM information. Furthermore, multisite features preserve information about phosphorylation sites that occur together on the same peptide molecule, revealing co-modification patterns that indicate coordinated regulation by kinases or functional relationships between sites. Traditional site-level analysis treats each phosphorylation site independently, losing this valuable information about which specific combinations of sites are modified together in biological samples.
Site-level reports collapse signals from all peptidoforms mapping to
the same PTM at an amino acid residue into a single quantitative value,
ensuring each site has exactly one intensity measurement per sample. In
downstream analysis, site-level intensities integrate seamlessly with
site-centric enrichment tools and kinase-activity inference platforms
(PhosR [28],
PTM-SEA [29], RoKAI
[30], etc.),
streamlining biological interpretation and network reconstruction
analyses [31].
Statistical Analysis of PTM Data
Statistical analysis of PTM data requires specialized methods due to
confounding effects between PTM levels and protein abundances. Several
statistical packages have emerged to address the critical issue of
integrating PTM-feature quantifications with protein abundance data.
Notable examples include MSstatsPTM [4] and
msqrob2PTM [5], which explicitly
model both modified peptide and protein-level changes to distinguish
genuine PTM regulation from protein abundance effects accurately.
The msqrob2PTM framework exemplifies the statistical
approaches now available for PTM analysis, defining two complementary
analytical strategies: DPA and DPU [5]. DPA directly
models the
intensities of each modified peptide (peptidoform), detecting absolute
changes in PTM levels between experimental conditions. In contrast, DPU
adjusts the PTM intensities by the corresponding protein-level changes,
effectively testing for changes in the relative usage or stoichiometry
of a modification site. This dual approach enables researchers to
distinguish between PTM changes driven by protein abundance differences
(DPA) versus those reflecting genuine changes in modification efficiency
or regulatory activity (DPU).
These two frameworks differ in their analytical approaches for
addressing confounding between PTM and changes in protein abundance.
MSstatsPTM employs a “model then correct” strategy,
separately fitting linear models to modified and unmodified peptide
data, and then combining statistical inferences post-modeling. In
contrast, msqrob2PTM follows a “correct then model”
approach, first adjusting PTM intensities by their corresponding protein
abundances at the data level, and subsequently performing statistical
testing on these normalized values.
A further option, discussed in [32], is to model the
PTM and total proteome data as a factorial design that jointly includes
a biologically relevant explanatory variable, e.g., genotype, and a
“component” indicator, i.e., modified peptide and its total proteome
parent protein, and fits a linear model with their interaction. It then
tests the statistical significance of the interaction term to flag
modified peptides whose dynamics deviate from the corresponding total
proteome protein abundance. Here, the package limma is used
to fit the model and perform the testing.
While all three methodologies ultimately estimate PTM abundance changes corrected for protein-level effects, each method can yield different test results due to varying assumptions about the relationship between PTMs and protein abundances within a sample, as well as differences in how variance and degrees of freedom are estimated.
-
MSstatsPTMfits the PTM and the protein in separate models, then computes an adjusted effect as a difference of estimated contrasts: with standard error combined in quadrature for the test statistic, and Welch’s approximation to estimate the degrees of freedom, which yields p-values for the adjusted fold change directly on the contrast scale.MSstatsPTMassumes that there is no correlation between PTM and the protein abundances within the samples. - The single-model approach [32] uses
limmamoderated (empirical-Bayes) variances for the interaction contrast within one fit, which shrinks feature-wise variances toward a common prior; are moderated accordingly. The single-model formulation can capture shared covariance by adding a sample blocking/random effect. Still, as illustrated [32], the baseline model omits a sample term, effectively treating PTM and protein measurements as independent. -
msqrob2PTMassumes that the PTM and the protein abundances within a sample are correlated and therefore corrects PTM abundances with the matched protein abundances. Then, for modelling, only the corrected PTM intensities are used.
The three pipelines target the PTM-vs-protein effect through different modeling paths and make different assumptions about within-sample covariance, and differ in variance moderation. Therefore, even with the same raw data and contrasts, we can expect differences in estimated effects, standard errors, and p-values.
To offer researchers additional flexibility, we developed
prophosqua [33], an R
package designed to streamline statistical analysis of phosphoproteomics
and other PTM datasets. Using prophosqua and the
prolfqua package [34], the
MStatsPTM model-first and msqrob2PTM
correct first approaches can be executed to obtain
protein-corrected PTM data and provide intuitive visualizations for
comprehensive exploratory analysis.
The Protein Assignment Problem in Differential PTM-feature Usage
Identification and quantification tools need to assign peptides to proteins, i.e., find the leading or representative protein identifier (ID). This assignment may be dataset-specific and may differ between the PTM and the total proteome dataset. Therefore, using protein IDs to match PTM features with proteins from the total run may lead to a loss of information due to differing representative IDs [35]. For example, a phosphopeptide might be assigned, by protein inference, to protein isoform A in the PTM dataset. In contrast, in the total proteome dataset, protein abundance is quantified under isoform B, which prevents proper integration and DPU analysis.
A robust solution involves matching stripped peptide sequences from the PTM dataset to peptide sequences of proteins quantified in the total proteome dataset, bypassing protein ID dependency entirely. When peptides are shared among multiple protein isoforms, the averaged protein abundance across all matching isoforms provides a more stable reference for normalization. While this sequence-based matching approach requires additional computational implementation, it reduces the loss of valuable information due to protein assignment differences between datasets.
Chapter Overview and Learning Objectives
This chapter introduces an analytical workflow that addresses the biological challenge of accurately distinguishing DPA from DPU. Distinguishing between protein-level effects and genuine modification-specific changes is critical for meaningful interpretation of PTM data. The workflow emphasizes reproducibility, automation, and biological interpretation.
DPA tests raw PTM signal changes between experimental conditions, identifying any modification abundance changes regardless of underlying protein-level effects. This analysis captures the total impact of experimental perturbations on PTM levels, including both direct modification effects and indirect effects mediated through changes in protein abundance.
DPU evaluates protein-normalized PTM changes, explicitly identifying modification sites where stoichiometry is genuinely altered independent of protein abundance. This analysis pinpoints sites where experimental conditions directly impact modification efficiency, kinase activity, or phosphatase activity, thereby highlighting specific regulatory events.
Learning objectives include: 1. Conducting integrated statistical
analysis of PTM and total proteome data using prolfquapp
and prophosqua R packages 2. Interpreting DPA
vs. DPU results for biological insights and generating
publication-quality visualizations 3. Performing sequence motif analysis
for kinase prediction and pathway reconstruction
Materials
Demonstration Dataset
This protocol utilizes the Atg16l1 macrophage dataset from Maculins
et al. [36] to demonstrate the
bioinformatics workflow and integrated statistical analysis. The dataset
comprises TMT-11-plex measurements from six conditions (WT/KO ×
uninfected/early/late infection) with both phospho-enriched and total
proteome samples, making it ideal for illustrating the principles of
integrated PTM analysis. In the original publication, the authors also
performed ubiquitin remnant enrichment, although we focus on the
phosphoproteome and total proteome datasets for demonstration purposes.
This same dataset was previously used in the MSstatsPTM
publication [4], providing
additional validation of the analytical approaches.
As discussed below, TMT enables high-precision quantification across many conditions, particularly when combined with fractionation. In contrast, DIA is well suited when avoiding fractionation or scaling to larger sample numbers (see Note @ref(notelfq)).
System Requirements
- Search and quantification software: FragPipe 22.0 (free download for academic use from fragpipe.nesvilab.org; system requirements: 32GB+ RAM, 50GB+ storage)
- Statistical platform: R version 4.0.0 (https://www.r-project.org/) and RStudio (https://posit.co/)
- System requirements: 16GB+ RAM, 20 GB+ storage
- Expected runtime: 10 minutes to 1 hours depending on dataset size and number of fractions
R packages
Most relevant R packages used for the analysis are:
-
tidyverse[37] - Data manipulation and visualization tools -
ggseqlogo[38] - Sequence motif visualization -
prolfqua[34] - Core statistical framework for proteomics data analysis -
prolfquapp[39] - Streamlined differential expression analysis pipeline -
prolfquappPTMreaders[40] - Specialized readers for PTM data formats -
prophosqua[33] - Integration and visualization of PTM and protein data
For detailed installation instructions, see the package documentation on
Mass Spectrometry Data Processing
Raw MS data acquired using DDA combined with TMT labeling are processed using specialized software to:
- Search spectra against a protein sequence database including species-specific sequences, common contaminants, and decoy sequences
- to extract reporter ion intensities
- and to compute site localization probabilities
The section (see Methods @ref(fragpipeparams)) describes in detail how we parameterized the FragPipe software [17].
Methods
A key challenge, when analysing PTMs, is to distinguish changes in
PTM abundance that are due to altered protein expression from those that
reflect a change in the modification stoichiometry (i.e., the fraction
of the protein pool that is modified). This protocol provides a
step-by-step computational workflow to analyze PTM data in the context
of total protein abundance changes. We leverage the R packages
prolfquapp [39] for streamlined
differential expression analysis [34] and
prophosqua [33] for the integration,
analysis, and visualization of PTM and total proteome data. The workflow
comprises three parts: 1) analysing the raw mass spectrometric data with
FragPipe, 2) performing differential expression analysis for
the PTM-enriched and total proteome datasets, and 3) integrating the PTM
and total proteome data.
Analysing TMT raw data with FragPipe
We used the TMT 11-plex phospho workflow in FragPipe 22.0 [17] with MSFragger (version 4.1) [23]. Database searching was performed against a species-specific protein sequence database supplemented with common contaminants and reversed decoy sequences. The search parameters included:
- Fixed modifications:
- Carbamidomethylation of cysteine (+57.0215 Da)
- TMT labeling of lysine residues and peptide N-termini (+229.1629 Da)
- Variable modifications:
- Phosphorylation on serine, threonine, and tyrosine residues (+79.9663 Da)
- Oxidation of methionine (+15.9949 Da)
- Acetylation at protein N-termini (+42.0106 Da)
Reporter ions are quantified by IonQuant (version 1.10.27), with downstream processing in TMTIntegrator (version 1.10.27). TMTIntegrator [22] performs quantification and normalization specifically tailored for multiplexed TMT experiments.
Selected TMTIntegrator parameters are:
- Best PSM selection: OFF
- Outlier removal enabled: OFF
- Allow overlabel/underlabel: OFF
- Use MS1 intensity: OFF
- Aggregation method: Median
For phospho-enriched samples, PTM sites are scored using
PTMProphet [24] localization
scores
0.75 are retained. FragPipe workflow parameters are stored in
configuration files (fragpipe.workflow). The workflow file
used to parametrize FragPipe can be downloaded from [41].
FragPipe provides two types of PTM-features reports for the phospho-enriched samples:
Multisite features: groups peptidoforms sharing identical modification sites, as described in Section @ref(data-analysis-frameworks).
Site-level reports collapse signals from all peptidoforms mapping to the same amino acid position into a single quantitative value per PTM site, ensuring each site has exactly one intensity per sample. In downstream analysis, site-level intensities can be fed into site-centric enrichment or kinase-activity inference tools (
PhosR,PTM-SEA,RoKAI, etc.), streamlining biological interpretation and network reconstruction.
An essential step in the data preprocessing is the quality control (QC) of the quantified samples, which involves estimating the missed cleavage rates or labeling efficiency (see Note @ref(automatedqc)).
Data Setup for Statistical Analysis
The following sections demonstrate the computational workflow through
R code snippets that implement each step of the integrated PTM
analysis. All code examples are also available within the
prophosqua package [33] and can be easily
adapted to different datasets by modifying file paths and experimental
parameters.
The first step is to download the example PTM dataset, which serves as a comprehensive demonstration of integrated PTM analysis. This dataset is derived from the Atg16l1 macrophage study by Maculins et al. [36], which investigates the role of autophagy in bacterial infection responses. The dataset contains both total proteome and PTM-enriched measurements from a well-designed factorial experiment with the following characteristics:
- Experimental design: factorial (Genotype: WT/KO; Timepoint: Uninfected/Early/Late infection)
- Biological context: Macrophage responses to Shigella infection in wild-type vs Atg16l1-knockout cells
- Technical approach: TMT 11-plex labeling for precise quantification across conditions
- Data types: Total proteome protein-level and phospho-enriched phospho-site quantification results.
This dataset is particularly valuable for demonstrating PTM analysis concepts because the Atg16l1 knockout creates both direct protein abundance changes (loss of Atg16l1 itself) and secondary effects on autophagy-related signaling pathways.
The first step involves obtaining the FragPipe 22 mass spectrometry output files [41] and setting up the analysis environment in R. Depending on the sample type, different FragPipe output files are required:
- total proteome samples:
psm.tsvfiles, containing peptide-spectrum match (PSM) information - phospho-enriched samples:
abundance_single-site.tsvfiles for individual modification site analysis
This dataset is available from Zenodo 10.5281/zenodo.15879865. The code below downloads the data, extracts it into a folder, and removes the zip archive.
need_download <- !dir.exists("PTM_example")
zenodo_url <- "https://zenodo.org/records/15879865/files/PTM_experiment_FP_22_Maculins_and_QC.zip"
destfile <- basename(zenodo_url)
# Download, unzip and delete
download.file(zenodo_url, destfile, mode = "wb")
unzip(destfile, exdir = ".")
unlink(destfile)Setting up prolfquapp scripts
This section demonstrates how to perform differential
expression analysis (DEA) on both total proteome and
PTM-enriched proteome data using the prolfquapp package.
The analysis provides the foundation for subsequent integration by
preprocessing, normalizing the data, and establishing which proteins and
PTM-features show significant changes between experimental
conditions.
The prolfquapp [39] package
provides a set of shell scripts that automate the analysis workflow.
These scripts are copied into the working directory by running the
command in the Linux shell and are used in subsequent steps.
Creating sample annotation
Sample annotation is the foundation of any meaningful DEA, serving as the bridge between raw mass spectrometry files and biological interpretation. The annotation file maps each raw data file to its corresponding experimental conditions (WT or KO) and additional explanatory variables, such as Time.
We start by reading the sample names from the FragPipe
output files using the prolfqua_dataset.sh script to create
a dataset annotation file compatible with prolfquapp, that
maps sample names to their experimental conditions (see Table
@ref(tab:showAnnotTable)).
./prolfqua_dataset.sh --indir PTM_example/data_total/FP_22/ \
--dataset "PTM_example/data_total/dataset.tsv" \
--software prolfquapp.FP_TMTWe use R to extract experimental factors, the Genotype and
Timepoint, from the channel labels using the
tidyr::separate function. If the channel names do not
encode the experimental conditions, the explanatory variables must be
provided by adding columns to the annotation file using a text
editor.
annot <- readr::read_tsv("PTM_example/data_total/dataset.tsv")
annot <- annot |> tidyr::separate("Name", c("Genotype", "Timepoint", NA),
sep = "_", remove = FALSE)
annot$group <- NULL
annot$subject <- NULL
annot$CONTROL <- NULLWe then call the annotation_add_contrasts function in
the prolfqua [34] to generate
factorial contrasts automatically. The
annotation_add_contrasts function adds two key columns to
the sample annotation:
-
ContrastName: A descriptive name for each statistical contrast -
Contrast: The mathematical formula defining the contrast in terms of group means
annot2 <- prolfqua::annotation_add_contrasts(
annot, primary_col = "Genotype", secondary_col = "Timepoint" ,
decreasing = TRUE, interactions = FALSE)$annot
write_tsv(annot2, "dataset_with_contrasts.tsv")Note that setting the argument interactions=FALSE in the
annotation_add_contrasts function disables the generation
of interaction contrasts for factor level combinations. Interaction
contrasts assess whether the effect of one factor depends on the level
of another, thereby capturing non‑additive interplay between factors.
The file dataset_with_contrasts.tsv is the final dataset
file that will be used for the DEA (see Table
@ref(tab:showannotation)).
For the experimental design in this protocol ( factorial: Genotype [KO/WT] Time [Uninfected/Early/Late]), the function generated four distinct contrasts:
-
KO_vs_WT(Main effect):( (G_KO_Uninfect + G_KO_Late + G_KO_Early)/3 - (G_WT_Uninfect + G_WT_Late + G_WT_Early)/3 )- Compares overall genotype effects across all timepoints -
KO_vs_WT_at_Uninfect:G_KO_Uninfect - G_WT_Uninfect- Compares genotypes specifically in the uninfected condition -
KO_vs_WT_at_Late:G_KO_Late - G_WT_Late- Compares genotypes specifically in the late infection condition
-
KO_vs_WT_at_Early:G_KO_Early - G_WT_Early- Compares genotypes specifically in the early infection condition
If we want to compare timepoints instead of genotypes, we set:
primary_col="Timepoint" and
secondary_col = "Genotype".
Execution of Differential Abundance Analysis
Before performing DEA, we generate a YAML configuration file using
the prolfqua_yaml.sh script. The configuration file
(config.yaml) defines key analysis parameters, including
the normalization method (variance stabilization normalization (VSN) by
default), protein summarization strategy (Tukey’s median polish by
default), and quality-filtering thresholds such as q-value cutoffs for
peptide or protein-level evidence.
Once the sample annotation and configuration files are prepared, DEA
is executed via the prolfqua_dea.sh script. This script
reads the experimental design (sample IDs, group labels, and contrasts)
alongside the configuration file. Single-site or protein intensities are
-transformed
and variance-stabilized before modeling, improving homoscedasticity
across conditions.
Linear models are fitted and contrasts are tested using the
prolfqua R-package’s contrast API. Rather than
imputing missing values, the model adjusts the degrees of freedom based
on the number of actual observations per feature, which naturally
down-weights features with high missingness and improves the reliability
of the fold-change and p-value estimates. Empirical Bayes variance
moderation is applied to stabilize variance estimates, particularly when
sample size is limited.
First, we run the DEA analysis of the total proteome quantification
results. The script uses FragPipe psm.tsv file as
input, aggregates reporter ions abundances to the peptide level, and
then estimates the protein abundances. The code below demonstrates how
the prolfqua_dea.sh shell script can be executed on the
Linux command line.
./prolfqua_dea.sh -i PTM_example/data_total/FP_22 \
-d dataset_with_contrasts.tsv \
-y config.yaml \
-w total_proteome \
-s prolfquapp.FP_TMTThe prolfqua_dea application expects the following
paramater:
-
-i: Specifies input directory with FragPipe results -
-d: Uses the annotated dataset file -
-y: Applies configuration settings in theconfig.yamlfile. -
-w: Sets the name of the work unit -
-s: Specifies software format (FragPipe TMT)
After running the DEA, the results are stored in a time-stamped
directory. This code constructs the path to the results directory using
the prolfquapp::zipdir_name() function, which follows the
standard naming convention for DEA results.
prolfquapp::zipdir_name("DEA", workunit_id = "total_proteome")## [1] "DEA_20260217_WUtotal_proteome_vsn"The DEA of the single-site output of enriched phosphoproteome
examines changes at individual modification sites and is performed by
running the code below. The analysis provides site-specific information
about which particular amino acids are modified under different
conditions. The abundance_single-site_None.tsv file
generated by FragPipe is used as input.
./prolfqua_dea.sh -i PTM_example/data_ptm/FP_22 \
-d dataset_with_contrasts.tsv \
-y config.yaml \
-w phospho_STY \
-s prolfquappPTMreaders.FP_singlesitePlease note that using the -s argument, we point to the
correct reader function in the prolfquappPTMreaders
R package to read the FragPipe single-site output.
For each data type, prolfquapp generates a comprehensive
suite of outputs:
- Dynamic HTML reports containing interactive PCA plots, boxplots, volcano plots, and heatmaps for quality control and exploratory analysis
- XLSX tables (DE_
.xlsx) summarizing fold changes, moderated t-statistics, raw p-values, and Benjamini–Hochberg–adjusted p-values - GSEA rank files (GSEA_
.rnk) for downstream gene-set enrichment analysis - SummarizedExperiment objects (
.rds) for seamless import into Shiny-based visualization tools
These results can be integrated using the prophosqua
package for combined analysis of total proteome and PTM data. However,
it is essential to note that there are scenarios where performing
differential PTM-feature abundance analysis is the only practical option
(see Note @ref(useofDPADPU)).
Integrated PTM Analysis using prophosqua
The prophosqua package [33] provides tools for
integrating and analyzing PTM data with total proteome measurements. It
enables researchers to distinguish between changes in protein abundance
and changes in the usage of modification sites. This section explains
how to load the results of the differential abundance analysis,
integrate the PTM and total proteome data, and determine differential
PTM-feature usage.
Differential expression results from prolfquapp,
provided in Excel format, for the total proteome and the multi- or
single-site PTM analyses are imported into R. These datasets
are then integrated by performing a left join on protein identifiers,
merging PTM-level statistics with the corresponding protein-level values
for each condition.
We start by defining the project name and analysis type.
library(prophosqua)
wu_id <- "CustomPhosphoAnalysis"
fgcz_project <- "PTM_analysis"
oid_fgcz <- "fgcz_project"
datetoday <- format(Sys.Date(), "%Y%m%d")
project_dir <- "."We also create a results directory to store the PTM/total proteome integration results:
descri <- wu_id
res_dir <- file.path(
project_dir,
paste0(fgcz_project, "_", datetoday, "_singlesite_", descri)
)
if (!dir.exists(res_dir)) {
dir.create(res_dir, recursive = TRUE)
}In the code below, we define the paths to the DEA results in the XLSX
files stored by prolfquapp. We need to specify the workunit
names and the date when the DEA analysis was run.
# Use today's date if running DEA, otherwise use existing results
datedea <- if(do_dea) datetoday else "20251214"
tot_file <- prophosqua::dea_xlsx_path(project_dir, "total_proteome", datedea)
ptm_file <- prophosqua::dea_xlsx_path(project_dir, "phospho_STY", datedea)
# Directory paths
stopifnot(file.exists(tot_file))
stopifnot(file.exists(ptm_file))Finally, we load the differential expression results into the R session.
required_cols <- c("protein_Id", "protein_length", "contrast")
tot_res <- prophosqua::load_and_preprocess_data(tot_file, required_cols)
tot_res <- prophosqua::filter_contaminants(tot_res)
phospho_res <- prophosqua::load_and_preprocess_data(ptm_file, required_cols)
phospho_res <- prophosqua::filter_contaminants(phospho_res)The data integration step is critical for enabling DPU analysis and represents one of the most important technical aspects of the workflow. This step joins phosphorylation site data with total protein data to create a unified dataset where each PTM-feature is linked to its corresponding protein-level measurements.
The left_join operation used here ensures that all
PTM-features are retained in the analysis, even if their parent proteins
were not detected in the total proteome experiment. This approach
maximizes the information content by allowing protein normalization
where possible, while also reporting PTM-features that cannot be mapped
to total proteome measurements.
join_column <- c(
"protein_Id" = "protein_Id",
"contrast" = "contrast",
"description" = "description",
"protein_length" = "protein_length"
)
suffix_a <- ".site"
suffix_b <- ".protein"
combined_site_prot <- dplyr::left_join(
phospho_res,
tot_res,
by = join_column,
suffix = c(suffix_a, suffix_b)
)We also need to check the match rate between PTM sites and proteins,
that is, the number of PTM sites that have a protein match divided by
the total number of PTM sites. Currently, prolfquapp
matches PTM and protein features using exact protein IDs, which
introduces a limitation: discrepancies in representative protein
identifiers between the PTM-enriched and total proteome datasets may
result in the loss of mapping information (see Note
@ref(matchrates)).
# Calculate match rate by contrast
match_rates <- combined_site_prot |>
group_by(contrast) |>
summarize(
total_sites = n(),
matched_sites = sum(!is.na(diff.protein)),
match_rate = round(matched_sites / total_sites * 100, 1)
)The Table @ref(tab:b5matchrate) shows the match rates for each contrast.
Visualizing PTM-feature Abundance (DPA)
DPA tests the raw PTM-feature signal change between conditions, without any correction for its parent protein’s expression level. This analysis is used to flag any PTM-feature whose abundance changes, even if the parent protein itself is also up- or down-regulated.
DPA results are visualized using N-to-C plots, generated by the
prophosqua [33]
n_to_c_expression_multicontrast function. These multi-panel
plots map both the
fold changes of individual modification sites along the primary sequence
of the protein and the
fold change of the protein abundance from the total proteome experiment
across all contrasts simultaneously. This representation provides a
clear visual summary of both site-specific and overall protein
expression changes across multiple experimental comparisons. The N-to-C
expression plot (see Figure @ref(fig:fig1Q64337)) shows that the protein
SQSTM1 (UniProt id Q64337) and the phosphorylation sites are upregulated
in the KO vs WT comparison across different timepoints.
Because generating N-to-C plots is computationally intensive, we
generate plots only for proteins with at least one significant
phosphorylation site in any contrast. The False Discovery Rate (FDR)
threshold for determining significance can be adjusted in the
n_to_c_expression_multicontrast or
n_to_c_usage_multicontrast functions, allowing researchers
to balance computational efficiency with comprehensive visualization
based on their specific considerations. When no significant PTM sites
are detected despite biological expectations, one possible reason is
insufficient statistical power, which can be mitigated by increasing the
sample size (see Note @ref(nosignificantPTM)).
plot_data <- n_to_c_expression_multicontrast(
combined_site_prot, FDR_threshold = 0.01,
max_plots = 100, include_proteins = c("Q64337"))The next code snippet exports
N-to-C expression plots to a PDF file for further analysis
and presentation (see also Note @ref(compperf) why we limit the number
of N-to-C plots written into a file).
Analysis of Differential PTM-feature Usage (DPU) - model-first
DPU tests the protein-normalized changes of PTM-features. This analysis is essential for determining whether the fraction of a protein that is modified at a specific site changes between conditions. It isolates changes in modification stoichiometry from changes in overall protein abundance.
In the model-first approach, DPU is calculated by subtracting the
total proteome protein
fold change from the PTM-feature’s
fold change. The associated p-values are recalculated using a method
that combines the variance from both the PTM and protein-level models,
as implemented in prophosqua [33]
test_diff function, or in the MSstatsPTM
R-package [4].
The analysis involves three key mathematical components:
- Effect Size Calculation ()
- Standard Error Propagation ()
- Degrees of Freedom Calculation ()
The differential PTM usage effect size is calculated as the difference between PTM and total proteome protein fold changes for each comparison:
This difference represents the protein-normalized change in PTM abundance, where:
- Positive values - indicate increased modification stoichiometry (higher fraction of protein is modified)
- Negative values - indicate decreased modification stoichiometry (lower fraction of protein is modified)
- Zero values - indicate no change in modification stoichiometry relative to protein expression
The combined standard error accounts for uncertainty in both PTM and protein measurements:
The error propagation ensures that the statistical significance of usage changes reflects uncertainty from both datasets, providing a more robust assessment of the reliability of the observed changes.
The effective degrees of freedom for the combined test are calculated using Welch’s approximation:
, which provides appropriate degrees of freedom. The Welch approximation is essential when the two datasets have different experimental designs or sample sizes.
Using these calculated parameters, the t-statistic is computed:
The p-value is determined using the t-distribution with degrees of freedom. Multiple testing correction (e.g., Benjamini-Hochberg FDR) is applied across all tested sites.
The test_diff function calculates the difference of
differences between PTM and total proteome data, revealing site-specific
usage changes.
combined_test_diff <- prophosqua::test_diff(phospho_res, tot_res, join_column = join_column)The PTM-feature usage
fold changes, p-values, and FDR estimates are added to the dataframe
combined_test_diff.
The DPU results are visualized using N-to-C plots generated by
prophosqua::n_to_c_usage_multicontrast, which display the
protein-normalized PTM-feature abundance changes across all contrasts
simultaneously and highlight sites with significant changes in usage.
These multi-panel plots reveal which sites are being used more or less
actively, independent of total protein changes, enabling comparison
across all experimental conditions.
plot_data_usage <- n_to_c_usage_multicontrast(
combined_test_diff, FDR_threshold = 0.05,
max_plots = 100, include_proteins = c("Q64337"))In the Atg16l1 macrophage dataset [36], SQSTM1 (uniprot id Q64337) protein levels are markedly increased in Atg16l1-knockout (KO) versus wild-type (WT), reflecting loss of autophagy-mediated turnover. Phosphosite analysis reveals that the raw phosphopeptide intensities at , , and are similarly elevated in cKO compared to WT (DPA), consistent with higher protein abundance (see Figure @ref(fig:fig1Q64337)). However, when applying DPU normalization, which subtracts the total proteome protein fold change from each site’s phospho fold change, these apparent phosphorylation increases are substantially reduced, indicating that most of the phosphosite abundance changes arise from protein accumulation rather than genuine changes in modification stoichiometry (see Figure @ref(fig:fig2usageQ64337)). This example highlights how DPU effectively isolates regulatory events on SQSTM1 from confounding protein-level effects.
Additionally, individual high-confidence DPA/DPU sites should be carefully inspected for technical quality, including verification of protein coverage and assessment of missing values across treatment groups (see Note @ref(qcrecommend)).
x1_usage <- which(grepl("Q64337", plot_data_usage$protein_Id))
fig2_plot <- plot_data_usage$plot[[x1_usage[1]]]When evaluating statistical significance in DPU analysis, it is essential to avoid overinterpreting results (see Note @ref(overinterpret)). Statistical significance does not guarantee biological relevance; effect sizes should be considered alongside FDR values when prioritizing sites for further investigation. Key findings should be validated through orthogonal methods to confirm biological relevance.
Protein PTM Integration Report Generation
The integrated analysis results are compiled into a comprehensive
HTML report using an R markdown template
_Overview_PhosphoAndIntegration_site.Rmd. The report
includes:
- Overview statistics on identified phosphorylation sites
- Interactive scatter plots comparing protein vs PTM-feature changes
- Volcano plots highlighting differential PTM-feature usage
The report includes data tables that allow searching for a specific protein and phosphorylation feature, and highlighting it in the scatter plot showing the total proteome protein as a function of the PTM-site , as well as in volcano plots. When analyzing the interactive reports, different combinations of DPA and DPU results provide distinct biological insights that guide interpretation. For instance, DPA+ only indicates changes driven primarily by protein abundance alterations, where PTM increases reflect higher protein levels rather than enhanced modification efficiency (see Note @ref(dpuvsdpa)).
To generate the HTML report, we begin by creating a report configuration that includes the project ID, order ID, and a work unit description to be displayed in the interactive HTML report.
drumm <- prolfquapp::make_DEA_config_R6(
PROJECTID = fgcz_project,
ORDERID = oid_fgcz,
WORKUNITID = descri
)
# Copy integration files
prophosqua::copy_phospho_integration()## [1] "/Users/wolski/projects/prophosqua/inst/PTM_example_analysis_v2/_Overview_PhosphoAndIntegration_site.Rmd"
## [2] "/Users/wolski/projects/prophosqua/inst/PTM_example_analysis_v2/bibliography2025.bib"Next, we can render the interactive HTML report with comprehensive analysis results, including tables, plots, and statistical summaries. This report is available for download from Zenodo [42].
# Render HTML report
rmarkdown::render(
"_Overview_PhosphoAndIntegration_site.Rmd",
params = list(
data = combined_test_diff,
grp = drumm
),
output_format = bookdown::html_document2(
toc = TRUE,
toc_float = TRUE
),
envir = new.env(parent = globalenv())
)
# Copy HTML report to results directory (uses existing if not re-rendered)
if (file.exists("_Overview_PhosphoAndIntegration_site.html")) {
file.copy(
from = "_Overview_PhosphoAndIntegration_site.html",
to = file.path(res_dir, "Result_phosphoAndTotalIntegration.html"),
overwrite = TRUE
)
}## [1] TRUEIn addition, the analysis results are exported to an Excel file for further analysis and sharing. The Excel file contains two worksheets:
- combinedSiteProteinData: Contains the merged PTM and protein-level data used for the DPA analysis.
- combinedStats: Contains the differential PTM-feature usage statistics (DPU), including fold changes, p-values, and FDR estimates for each phosphorylation site.
This Excel format facilitates downstream analysis, data sharing with collaborators, and integration with other bioinformatics tools. The file is automatically saved with a timestamped filename in the results directory.
excel_result_list <- list(
combinedStats = combined_test_diff,
combinedSiteProteinData = combined_site_prot
)
writexl::write_xlsx(
excel_result_list,
path = file.path(
res_dir,
"Result_phosphoAndTotalIntegration.xlsx"
)
)
# Save example data for package (as .rda for data() loading)
combined_test_diff_example <- combined_test_diff
save(combined_test_diff_example,
file = here::here("data", "combined_test_diff_example.rda"),
compress = "xz")The produced XLSX file can be downloaded from [42]. This comprehensive integrated analysis framework provides key advantages for PTM research, including the ability to distinguish genuine signaling changes from protein abundance effects (see Note @ref(keyinsight)).
Estimating PTM-feature usage by the correct first approach
Here we show how to estimate the DPU using the correct first
approach. We first correct the PTM site abundances, using the total
proteome, and then model the differential usage changes. This analysis
starts by loading the prolfquapp outputs. This time, we
read the transformed and normalized total proteome protein
abundances.
tot_res_dir <- prophosqua::dea_res_dir(project_dir, "total_proteome", datedea)
ldata <- arrow::read_parquet(file.path(tot_res_dir,"lfqdata_normalized.parquet"))
ldata <- ldata |> filter(!grepl("^rev_",protein_Id) )
tot_d <- ldata |>
dplyr::select(channel, protein_Id, normalized_abundance)We are also loading normalized single-site abundances. In addition to
loading the data, we create a prolfqua::LFQData object
using the LFQData$new constructor. The LFQData
object provides numerous methods for analyzing and visualizing
quantitative proteomics data [34].
ptm_res_dir <- prophosqua::dea_res_dir(project_dir, "phospho_STY", datedea)
ldata <- arrow::read_parquet(file.path(ptm_res_dir,"lfqdata_normalized.parquet"))
list <- yaml::read_yaml(file.path(ptm_res_dir,"lfqdata.yaml"))
config <- prolfqua::list_to_AnalysisConfiguration(list)
ptm_data <- prolfqua::LFQData$new(ldata,config)We merge the data for the single-site and total proteome protein abundances by protein identifier and sample. Then we subtract the total proteome protein abundances from the single-site abundances. The Density plot (see Figure @ref(fig:fig3densityCorrected)) shows the distribution of the corrected single-site abundances.
ptm_data$data <- dplyr::inner_join(
ptm_data$data, tot_d,
by = c("channel", "protein_Id"), suffix = c(".site",".total"))
ptm_data$data <- ptm_data$data |>
dplyr::mutate(ptm_usage = normalized_abundance.site - normalized_abundance.total)
ptm_data$config$table$set_response("ptm_usage")
pl <- ptm_data$get_Plotter()
fig3_plot <- pl$intensity_distribution_density()Next, we fit a linear model for each PTM, which explains the
corrected PTM site abundances using the grouping of the samples. The
model is fitted for each site using the
prolfqua::build_model function.
strategy_lm <- prolfqua::strategy_lm("ptm_usage ~ G_")
models <- prolfqua::build_model(data = ptm_data, model_strategy = strategy_lm)To compute the contrasts, we construct an array of contrasts from the annotation table, which we built in Section @ref(contrasts).
contrasts <- annot2$Contrast
names(contrasts) <- annot2$ContrastName
contrasts <- contrasts[!is.na(contrasts)]The following code fragment computes contrasts from the linear
models. In the presence of missing values, some PTMs cannot be fitted by
the model. Therefore, we also estimate contrast using the
ContrastsMissing method, which estimates missing values
based on the inferred limit of detection (for more details, see [34]).
ctr <- prolfqua::Contrasts$new(models, contrasts)
x <- ctr$get_contrasts()
ctr_m <- prolfqua::ContrastsMissing$new(ptm_data, contrasts)
ctr_all <- prolfqua::merge_contrasts_results(ctr, ctr_m)$merged
ctr_mod <- prolfqua::ContrastsModerated$new(ctr_all)
ctr_df <- ctr_mod$get_contrasts()Finally, we can create a volcano plot of the DPU results using the
ctr_plotter variable, which is an instance of the
prolfqua::ContrastsPlotter class (see Figure
@ref(fig:fig4FDRcorrfirst)).
ctr_plotter <- ctr_all$get_Plotter()
fig4_plot <- ctr_plotter$volcano()$FDRTo enable downstream analyses such as sequence motif analysis and
PTM-SEA, we need to enrich the contrast results with sequence window
information. The phospho_res dataframe loaded earlier
contains all the annotation data we need.
# Select relevant columns from phospho_res for joining
site_annotations <- phospho_res |>
dplyr::select(
site_Id = protein_Id,
SequenceWindow,
posInProtein,
modAA,
description,
protein_length
) |>
dplyr::distinct()
# Join to contrast results
ctr_df_annotated <- ctr_df |>
dplyr::left_join(site_annotations, by = c("modelName" = "site_Id"))
# Rename columns to match the model-first output format
ctr_df_annotated <- ctr_df_annotated |>
dplyr::rename(
diff_diff = diff,
FDR_I = FDR
)
# Save correct-first results alongside model-first output
saveRDS(ctr_df_annotated,
file.path(res_dir, "ctr_df_annotated_correctfirst.rds"))The ctr_df_annotated dataframe now contains both the DPU
statistics
(
fold changes, p-values, FDR) and the sequence annotations needed for
motif analysis and kinase activity inference.
Sequence Logo Analysis of Modified Sites
This section guides the biological interpretation of PTM changes by analyzing sequence motifs surrounding significantly modified sites to identify potential kinase recognition patterns. This analysis bridges the gap between statistical significance and biological mechanism (see Note @ref(overinterpret)) by leveraging the well-established principle that kinases recognize specific amino acid sequence patterns around their target sites.
Protein kinases exhibit sequence specificity, preferentially phosphorylating sites that match their recognition motifs. These motifs typically encompass positions -5 to +4 relative to the phosphorylation site, with specific positions being more critical than others. By analyzing the sequence patterns of significantly regulated sites, we can:
- Identify active kinases: Sites that share similar motifs likely represent targets of the same kinase or kinase family. Table @ref(tab:tablesites) shows examples of kinase-specific sequence motives.
- Understand pathway activation: Different kinase families are associated with different signaling pathways
- Predict functional consequences: Kinase-specific phosphorylation often has predictable effects on protein function
- Generate testable hypotheses: Motif predictions can guide follow-up experiments with kinase inhibitors or knockdowns
To identify potential kinases responsible for the observed
phosphorylation changes, sequence motif analysis is performed on
significantly regulated PTM sites identified in the DPU analysis. For
each contrast, amino acid sequences flanking the significantly regulated
sites (i.e.,
and
)
are extracted and grouped by regulation status (i.e., upregulated or
downregulated). The ggseqlogo R package is then
used to generate sequence logo plots for each group. These plots
visualize conserved amino acid patterns around the modification site,
facilitating comparison to known kinase recognition motifs and enabling
inference of upstream regulatory kinases (see Table
@ref(tab:tablesites)).
The analysis focuses on sites with strong statistical evidence to ensure that motif patterns reflect genuine biological changes rather than technical artifacts. For this dataset, this stringent filtering yields many sites (see Table @ref(tab:tableSignificantContrasts)). However, for other datasets, the number of sites might be insufficient for creating sequence logos (see Note @ref(nosignificantPTM)).
We start by filtering the DPU data by and . The Table @ref(tab:tableSignificantContrasts) summarizes the number of PTM-features for each contrast, modification site, and up- or down-regulation. We observe that for serine (S) and threonine (T), there are many up- and downregulated sites, while for tyrosine (Y), there are only a few.
log_2FC_threshold <- 0.58
significant_sites <- combined_test_diff |>
dplyr::filter(
FDR_I < 0.05,
abs(diff_diff) > log_2FC_threshold,
!is.na(posInProtein),
!grepl("^_", SequenceWindow),
!grepl("_$", SequenceWindow)
) |>
dplyr::mutate(
regulation = dplyr::case_when(
diff_diff > 0 ~ "upregulated",
diff_diff < 0 ~ "downregulated",
TRUE ~ "no_change"
)
)Before generating sequence logos, we must ensure that the sequence windows are properly centered on the phosphorylation site. This code validates that the 8th character (center position) of each sequence window matches the reported modified amino acid (Note @ref(seqwindow)).
significant_sites$seventh_chars <- toupper(substr(significant_sites$SequenceWindow, 8, 8))
significant_sites <- significant_sites |> filter(seventh_chars == modAA)We generate sequence logos for sites of significantly regulated serine and threonine phosphorylation. The plots are organized by contrast in a 3-column layout showing upregulated sites, downregulated sites, and their difference (Up - Down). The difference column highlights amino acids that are enriched in upregulated sites (positive, above baseline) versus downregulated sites (negative, below baseline), helping to identify kinase-specific motifs (see Figure @ref(fig:fig5seqlogoST)).
sig_sites_ST <- significant_sites |>
dplyr::filter(modAA == "S" | modAA == "T")
fig5_plot <- plot_seqlogo_with_diff(sig_sites_ST)This analysis helps identify the signaling pathways and kinases responsible for the observed changes in phosphorylation under your experimental conditions. When motif analysis yields weak or absent sequence patterns, several factors may require optimization, including the number of significant sites, the sequence window size used for motif extraction (see Note @ref(motiflimit)).
Kinase Activity Inference Using ptm-pipeline
Kinase activity can be inferred from phosphoproteomics data by testing whether known or predicted substrates of a kinase are coordinately shifted in a ranked list of regulated phosphosites. Two complementary resources for defining kinase–substrate sets are (i) curated signatures from PTMsigDB (distributed via MSigDB’s PTMsig collection) and (ii) motif specificity models from The Kinase Library.
PTMsigDB / MSigDB PTMsig collection. The PTMsig collection (PTMsigDB) provides literature-curated, PTM site–specific signature sets for site-centric enrichment analysis, including kinase–substrate signatures, perturbation signatures (e.g., growth factor or inhibitor treatment), and pathway-related site signatures. Here, a “signature” refers to a named set of many phosphosites (analogous to a gene set in classic GSEA), not to a single sequence window. To maximize compatibility across search engines and protein database versions, PTMsigDB distributes signatures using multiple site identifier schemes, most commonly phosphosite-centered flanking sequences (typically amino acids around the modified residue; 15-mers) as well as UniProt-centric residue identifiers (and optionally PhosphoSitePlus site-group IDs). PTMsigDB provides organism-specific releases for human, mouse, and rat; in the original PTMsigDB publication, the total number of available signature sets was on the order of hundreds (reported as ), and counts vary by database version and organism.
The Kinase Library. A more comprehensive, model-based resource is The Kinase Library [43], which profiled substrate sequence preferences for 303 human Ser/Thr kinases (covering >84% of those predicted to be active) and provides position-specific scoring matrices (PSSMs). Unlike curated substrate lists that require exact matches to previously observed sites, PSSMs assign a quantitative compatibility score to any observed sequence window, enabling kinase–substrate relationships to be inferred without perfect matching. This makes it straightforward to construct “kinase substrate sets” by applying a score threshold or selecting the top-ranked sites per kinase, which can then be used as input signatures for enrichment testing.
PTM Signature Enrichment Analysis (PTM-SEA) using GSEA. Once kinase–substrate sets are defined (from PTMsigDB, motif/PSSM scoring, or both), kinase activity can be inferred quantitatively using PTM-SEA [29], which adapts the Gene Set Enrichment Analysis (GSEA) framework [44] to site-centric data. Phosphosites (or their sequence-window identifiers) are ranked by a differential signal (e.g., fold change or a t-statistic). GSEA then tests whether members of a kinase’s substrate set tend to occur near the top or bottom of this ranked list more than expected under a null model of uniform distribution. The enrichment score is computed by walking down the ranked list, increasing a running sum when a set member is encountered and decreasing it otherwise. Positive enrichment is consistent with increased kinase activity (substrates preferentially upregulated), while negative enrichment is consistent with reduced activity.
Practical considerations and coverage. PTMsigDB signatures provide high-confidence, literature-supported kinase assignments, but they are constrained by database coverage and identifier matching (including the need for consistent site definitions and sequence context). These constraints become more pronounced outside human (and to a lesser extent mouse/rat). In contrast, motif-based scanning (e.g., basophilic motifs such as R-x-x-S/T for PKA/CaMKII-like kinases) and Kinase Library PSSMs generalize to any observed sequence window, enabling broader application across datasets where curated substrate evidence is sparse—while trading off direct experimental validation at the individual site level.
In ptm-pipeline [45], kinase activity
inference is implemented as an automated workflow that uses
prolfquapp/prophosqua to generate differential
phosphorylation abundance (DPA) and differential phosphorylation usage
(DPU) results under both the “model” and “correct-first” strategies. It
then performs GSEA-based PTM-SEA using kinase substrate signatures from
PTMsigDB as well as Kinase Library–derived sets generated by PSSM
scoring of phosphosite sequence windows. The workflow is provided as a
command-line tool and executed via a Snakemake pipeline. The
source code is available at github.com/wolski/ptm-pipeline.
Notes
Label-free quantification vs. TMT-based approach
TMT-based quantification offers several advantages, including higher throughput, reduced missing values, and improved quantitative precision, while also enabling efficient use of fractionation since all samples are represented within each fraction. In contrast, label-free approaches avoid labeling-related artifacts, can handle an unlimited number of samples, and typically have lower per-sample costs. Fractionation strategies benefit particularly from TMT, as all samples can be compared directly within each fraction with reliable cross-fraction integration. In contrast, data-independent acquisition (DIA) is often preferred when avoiding fractionation altogether. Overall, we recommend TMT for studies requiring high precision across multiple conditions, particularly when fractionation can be leveraged to increase depth. In contrast, label-free or DIA workflows are well-suited for discovery-phase studies or experiments where the number of samples exceeds the multiplexing capacity.
Automated QC reporting with FragPipe TMT QC script
The prophosqua[33] package vignette
QCReport.qmd can be used to generate a comprehensive TMT
labeling efficiency report, directly from FragPipe PSM
psm.tsv output file. The script evaluates labeling
completeness at both peptide and PSM levels for N-terminal and lysine
modifications, calculates missed cleavage rates for tryptic digestion
efficiency, and provides quantitative balance assessment across all TMT
channels. Key metrics include the percentage of modified N-termini and
lysine residues (expected to be
),
missed cleavage rates for lysine and arginine residues, and relative
abundance distributions across channels to detect loading imbalances. To
assess labeling efficiency, the TMT labeling of lysine residues and
peptide N-termini must be specified as a variable modification in the
search.
Use of the integrated DPA/DPU approach
The integrated DPA/DPU approach is most suitable when the biological system under study exhibits substantial changes in protein expression. In such cases, distinguishing between signaling effects and expression-driven effects is essential, and including protein abundance measurements as part of the study design allows these two sources of variation to be effectively separated.
In contrast, enriched phosphoproteome measurements without corresponding protein data can be considered when protein expression is expected to remain stable across conditions, for example, in experiments with very short timescales where transcriptional or translational changes are unlikely. This strategy may also be justified when budget limitations make comprehensive protein quantification infeasible.
Match Rates
When evaluating integration between PTM and protein datasets, match rates above generally indicate good consistency. In contrast, lower rates may reflect differences in protein assignment between the PTM-enriched and total proteome analyses. In cases where match rates fall below , the underlying cause is often inconsistent protein identifiers between the two datasets. To address this, it is essential to verify that both analyses utilized the same FASTA database and identical protein inference settings, with particular attention to database versions and the inclusion of contaminant sequences. As a preventative measure, identical search parameters and databases should always be applied to both total proteome and PTM analyses, while sequence-based matching can further improve alignment across datasets.
No significant PTM sites detected despite biological expectation
The absence of significant PTM sites, even when biological regulation
is expected, can arise from several factors. Error propagation from both
PTM and protein measurements may amplify overall uncertainty, masking
accurate signals. Insufficient statistical power is another common
cause, emphasizing the need for at least three biological replicates per
group and, where possible, increasing sample size or reducing technical
variability. In some cases, stringent statistical thresholds may obscure
true positives, and adopting a more permissive cutoff (e.g., FDR = 0.25
instead of 0.05) during exploratory analysis can be informative.
Normalization problems, such as batch effects or inconsistent sample
loading, may also contribute and should be checked carefully. Outliers
in the data can further distort inference, making it useful to inspect
density or scatter plots, for example, in prolfquapp
reports. Finally, examining the distribution of p-values in
quality-control summaries can help assess whether the statistical model
adequately explains the variance in the data.
When no significant differential peptidoform usage (DPU) sites are detected despite clear changes in PTMs, one possible explanation is that the PTM changes are proportional to changes in the parent protein, indicating no alteration in modification stoichiometry. In many cases, this represents a valid biological result rather than a technical issue, and it is advisable to verify it by inspecting individual examples where protein and PTM changes are closely correlated. Alternatively, such an outcome may suggest that the experimental perturbation primarily influences protein expression rather than signaling activity.
Computational Performance Issues
Long processing times for N-to-C plots often occur because generating individual plots for hundreds of proteins is computationally intensive. A practical solution is to limit plot generation to proteins of particular interest, such as those with small FDR values and substantial fold changes, rather than producing plots for all detected proteins.
Quality Control Recommendations
When focusing on specific phosphorylation sites, it is essential to
validate key findings by carefully inspecting high-confidence DPU sites
to ensure both technical and biological plausibility. For example, this
can be achieved by verifying the number of missing values in each
treatment group. In addition, protein coverage should be verified, since
meaningful interpretation of phosphorylation changes requires that the
corresponding proteins have sufficient coverage in the total proteome.
The number of peptides identified per protein, as reported in the
prolfquapp output, can serve as a filtering criterion.
Overinterpretation of statistical significance
Overinterpretation of statistical significance can occur when FDR estimates are reported without considering the design of the experiment or its biological context. A statistically significant finding does not necessarily imply biological relevance; therefore, it is essential to evaluate effect sizes alongside p-values to assess the magnitude and potential impact of a change. Key findings should also be validated with orthogonal methods to ensure that they reflect genuine biological regulation rather than statistical or technical artifacts.
DPA vs DPU Interpretation
Conflicting results between DPA and DPU analyses can be due to:
- DPA+ only: Changes driven by protein abundance alterations
- DPU+ only: True signaling changes with stable protein levels
- DPA+ and DPU+: Amplified signaling (both protein and modification efficiency increase)
- DPA- and DPU+: Compensatory regulation (modification increases despite protein decrease)
Such discrepancies between DPA and DPU are often informative rather than problematic. Sites that are significant in DPA but not in DPU typically reflect protein-driven changes, whereas DPU-only sites indicate genuine signaling regulation independent of protein abundance. To assess their biological plausibility, these results should be cross-referenced with known biology and supporting literature.
Key Insights from This Analysis
This integrated analysis distinguishes PTM feature abundance from usage changes, enabling site-specific insights into how phosphorylation and other PTMs are regulated. By linking DPA and DPU results, it clarifies whether observed changes reflect protein-level expression or signaling pathway activity, and it highlights key regulatory sites for functional validation. Visualization in sequence context and rigorous statistical modeling further support interpretation, allowing researchers to generate mechanistic hypotheses, identify pathway-level regulation, and prioritize targeted follow-up experiments.
Extracting sequence windows with prophosqua
Although FragPipe reports the sequence windows in the
single-site report, we observed that some of the extracted windows are
not correctly aligned, making the filtering step necessary. Some
upstream quantification applications only report the modified amino acid
and the position in the protein sequence. For those cases, the
prophosqua function get_sequence_windows can
be used. The function expects a dataframe with two columns: the protein
sequences and the position of the modified amino acid. For details, see
prophosqua documentation [33].
Motif Analysis Limitations
Weak or absent sequence motifs in significantly regulated sites can arise for several reasons. One possibility is that too few significant sites were detected, which limits the statistical power of motif discovery. In such cases, it can be helpful to lower the threshold specifically for motif analysis to increase the number of sites included. Mixed kinase activities may also obscure motifs, and separating upregulated from downregulated sites often clarifies distinct regulatory patterns. Additionally, regulation may occur through non-canonical mechanisms, such as phosphatases or protein–protein interactions, rather than through specific kinase recognition motifs. Other technical considerations, such as the choice of sequence window size (e.g., versus amino acids), and biological factors like PTM crosstalk, can also influence motif detection and interpretation.
Overall, weak motif signals can result from an insufficient number of significant sites, the presence of multiple overlapping regulatory activities, or regulation by mechanisms that do not rely on canonical sequence motifs. To address these issues, one can lower the significance threshold for motif analysis to capture more sites. If no clear kinase sequence pattern emerges, keep in mind that regulation may involve non-kinase pathways, which underscores the importance of considering both statistical and biological explanations when interpreting motif analysis results.
References
Figures
(ref:fig1Q64337) Multi-panel N-to-C expression plot showing differential expression of total protein abundance (light yellow rectangle) and phosphorylation sites (vertical lines, color coded by type of residue) along protein sequences across all contrasts. Each panel represents a different experimental contrast, enabling direct comparison of protein and PTM expression changes across conditions. The vertical lines are either dashed or solid, depending on the site’s imputation status. The x-axis represents amino acid position from N to C terminus, while the y-axis shows fold changes. Significantly regulated sites (FDR < 0.05 and FDR < 0.01) are highlighted with one or two red asterisks, respectively.
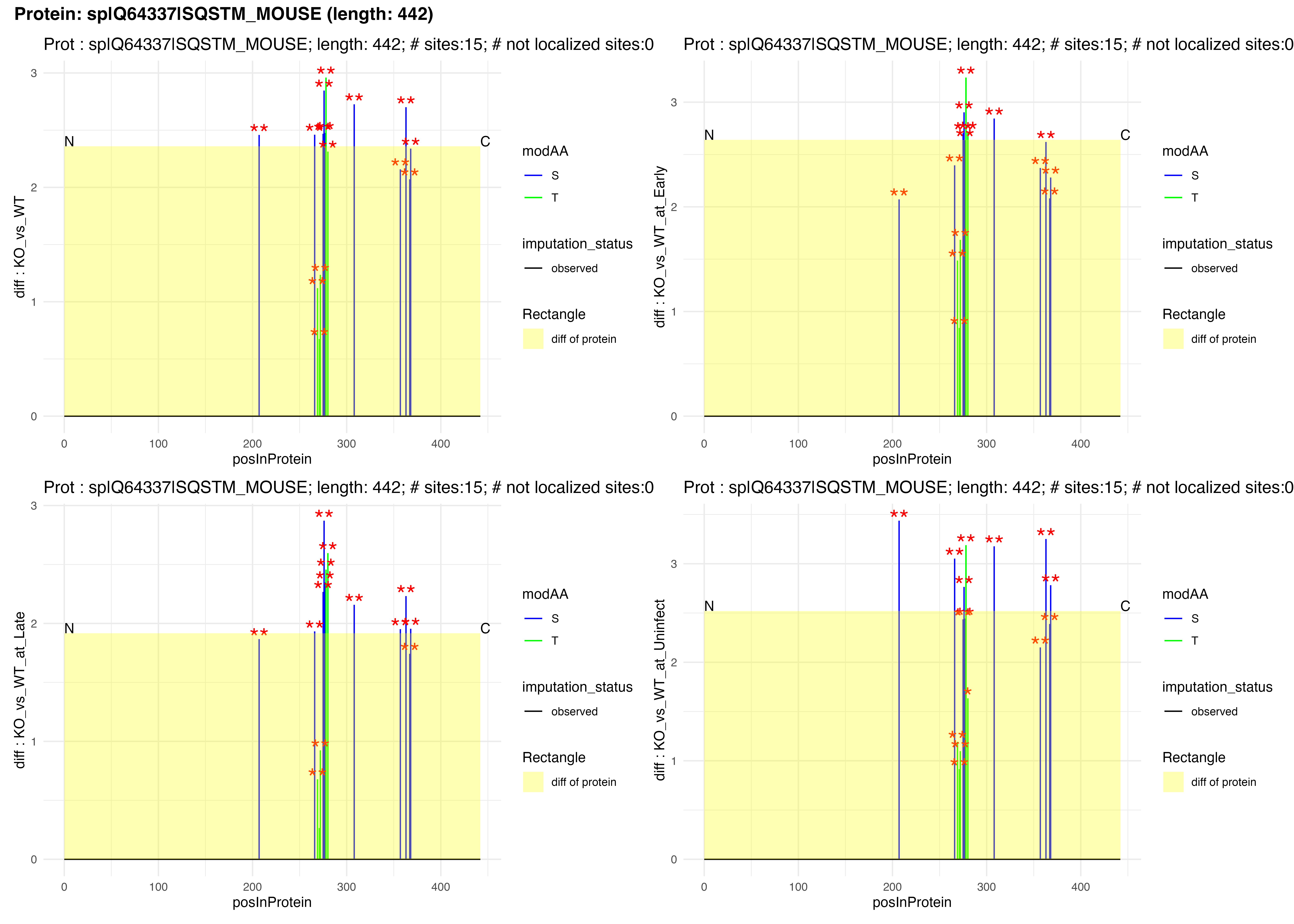
(ref:fig1Q64337)
(ref:fig2usageQ64337) Multi-panel N-to-C usage plot showing differential usage of phosphorylation sites (vertical lines, color-coded by type of residue) along the protein sequences across all contrasts. Each panel represents a different experimental contrast, enabling direct comparison of protein-normalized PTM changes across conditions. The vertical lines are either dashed or solid, depending on the site’s imputation status. The x-axis represents amino acid position from N to C terminus, while the y-axis shows fold changes. Significantly regulated sites (FDR < 0.05 and FDR < 0.01) are highlighted with one or two red asterisks, respectively.
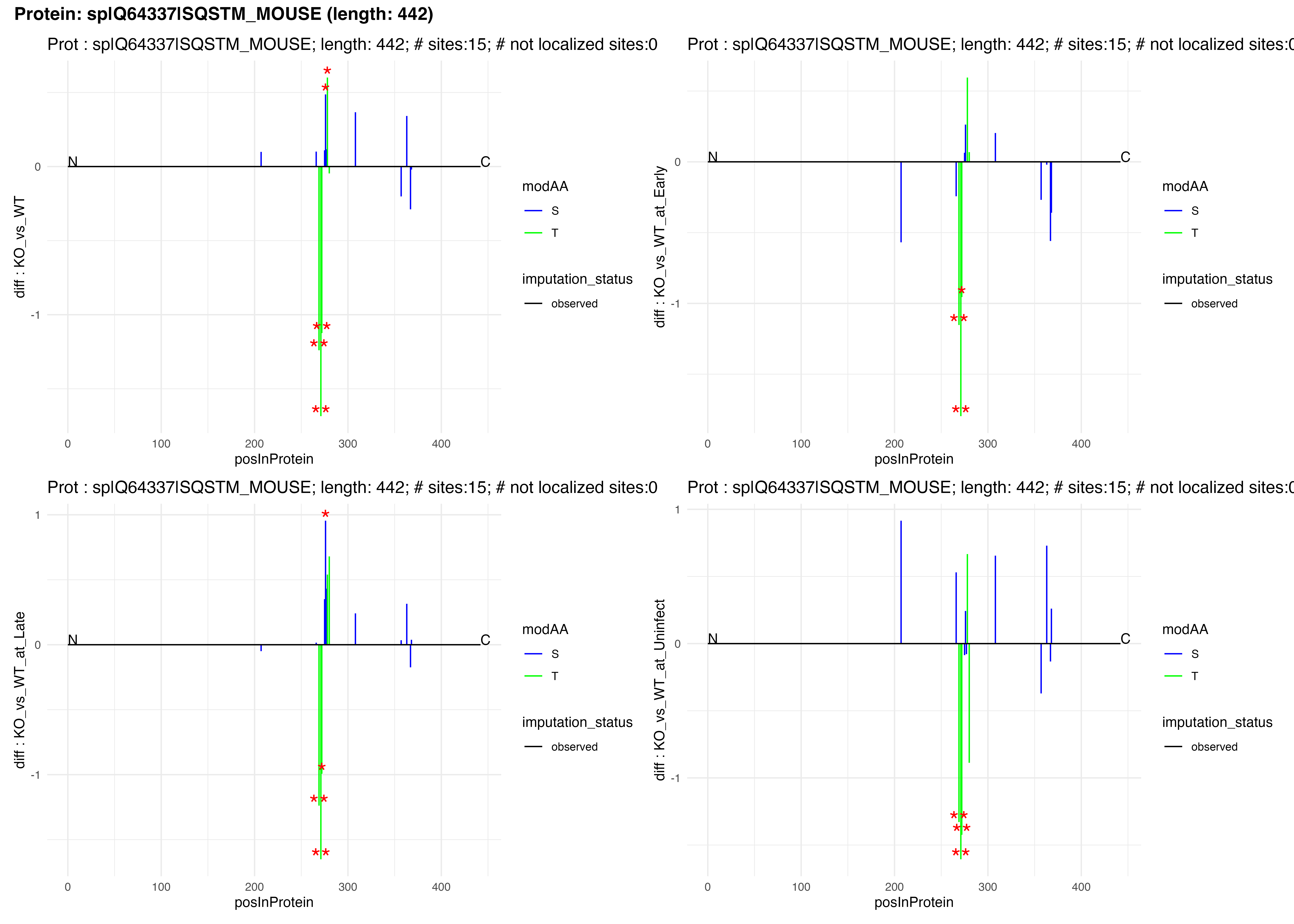
(ref:fig2usageQ64337)
(ref:fig3densityCorrected) Density plot of the total proteome corrected PTM site abundances.
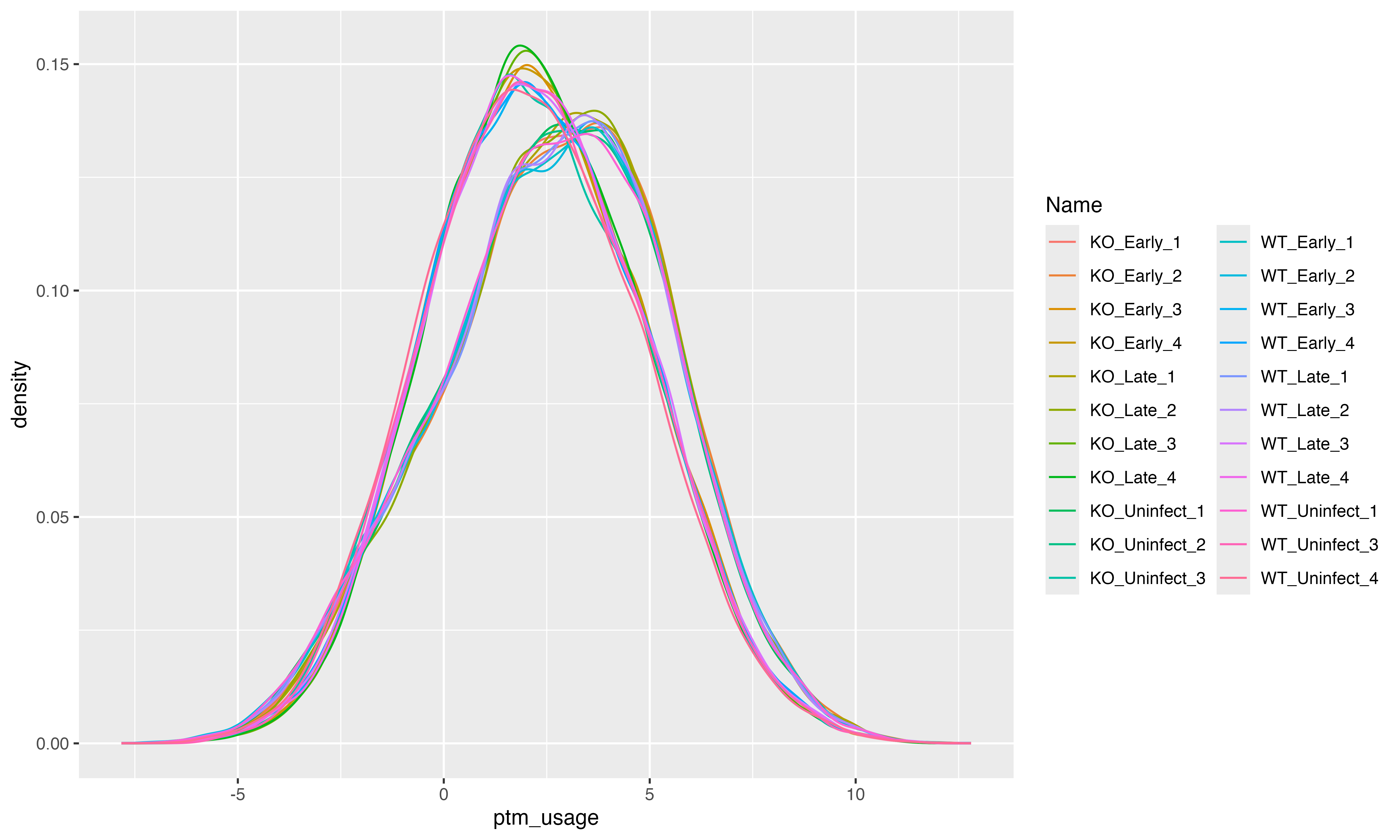
(ref:fig3densityCorrected)
(ref:fig4FDRcorrfirst) Volcano plot showing the as a function of the PTM site fold change.
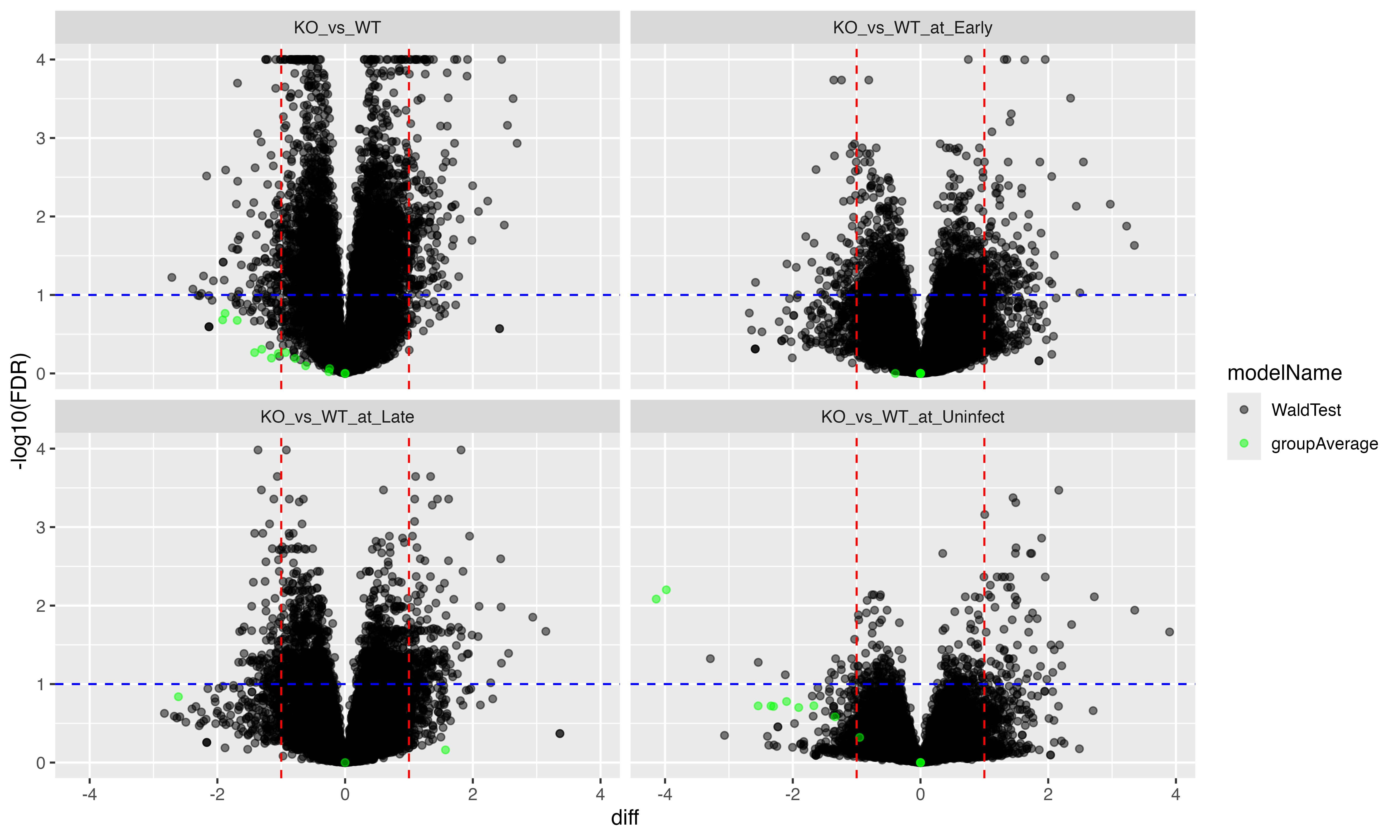
(ref:fig4FDRcorrfirst)
(ref:fig5seqlogoST) Sequence logo plots showing amino acid motifs surrounding significantly regulated phosphorylation at serine and threonine residues. Each row represents a contrast with three columns: upregulated sites (left), downregulated sites (center), and the difference (right). In the difference column, letters above the baseline indicate amino acids enriched in upregulated sites, while letters below the baseline indicate enrichment in downregulated sites. Letter height corresponds to relative frequency at each position surrounding the phosphorylation site (position 8).
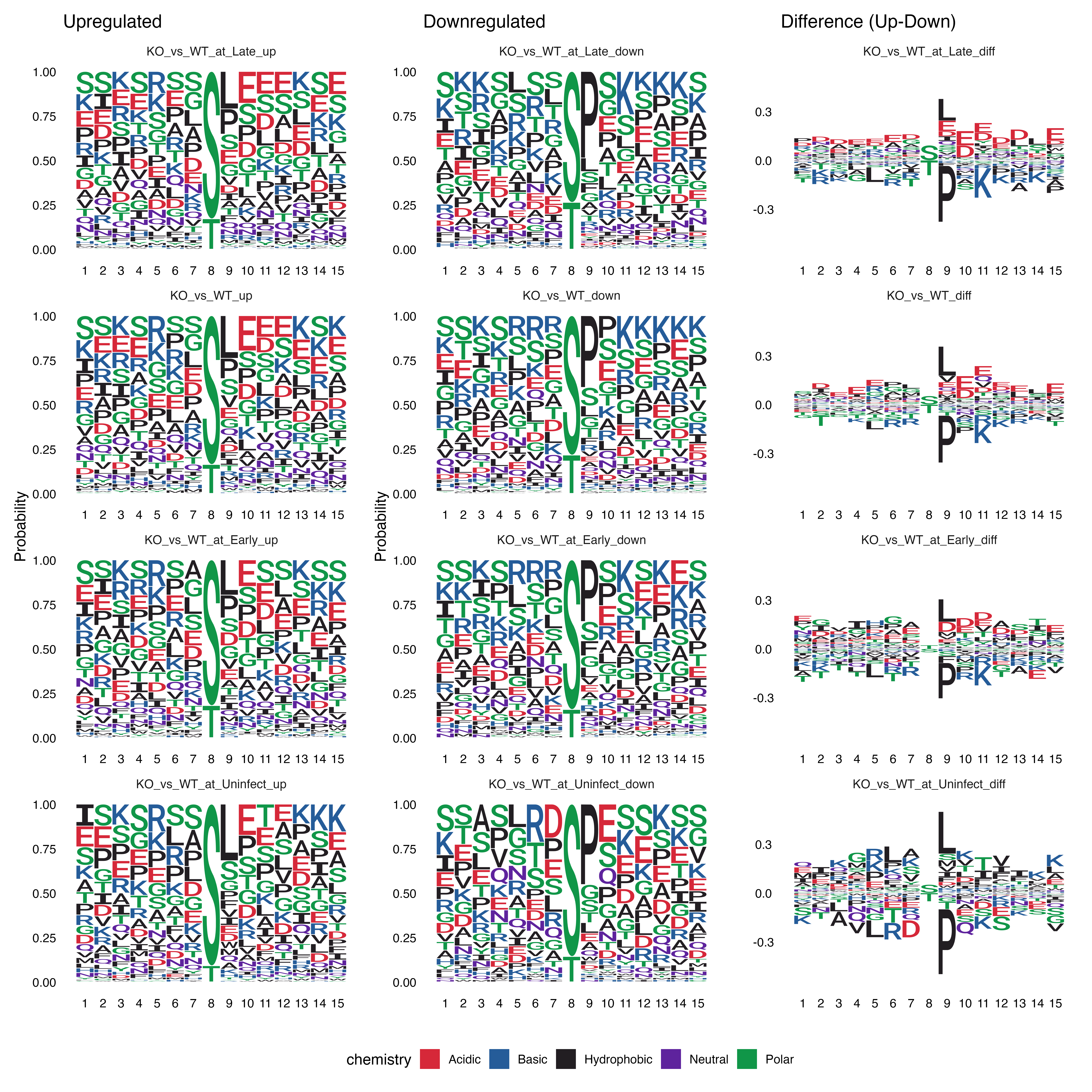
(ref:fig5seqlogoST)
Tables
| channel | Name | group | subject | CONTROL |
|---|---|---|---|---|
| KO_Early_1 | KO_Early_1 | NA | NA | NA |
| KO_Early_2 | KO_Early_2 | NA | NA | NA |
| KO_Late_1 | KO_Late_1 | NA | NA | NA |
| KO_Late_2 | KO_Late_2 | NA | NA | NA |
| KO_Uninfect_1 | KO_Uninfect_1 | NA | NA | NA |
| KO_Uninfect_2 | KO_Uninfect_2 | NA | NA | NA |
| channel | Name | Group | ContrastName | Contrast |
|---|---|---|---|---|
| KO_Early_1 | KO_Early_1 | KO_Early | KO_vs_WT | ( (G_KO_Uninfect + G_KO_Late + G_KO_Early)/3 - (G_WT_Uninfect + G_WT_Late + G_WT_Early)/3 ) |
| KO_Early_2 | KO_Early_2 | KO_Early | KO_vs_WT_at_Uninfect | G_KO_Uninfect - G_WT_Uninfect |
| KO_Late_1 | KO_Late_1 | KO_Late | KO_vs_WT_at_Late | G_KO_Late - G_WT_Late |
| KO_Late_2 | KO_Late_2 | KO_Late | KO_vs_WT_at_Early | G_KO_Early - G_WT_Early |
| KO_Uninfect_1 | KO_Uninfect_1 | KO_Uninfect | NA | NA |
| KO_Uninfect_2 | KO_Uninfect_2 | KO_Uninfect | NA | NA |
| Contrast | Total Sites | Matched Sites | Match Rate |
|---|---|---|---|
| KO_vs_WT | 21733 | 21000 | 96.6 |
| KO_vs_WT_at_Early | 21733 | 21000 | 96.6 |
| KO_vs_WT_at_Late | 21733 | 21000 | 96.6 |
| KO_vs_WT_at_Uninfect | 21733 | 21000 | 96.6 |
| Kinase | Consensus_motif | Positional_notes |
|---|---|---|
| PKA/PKG | R-R-X-S/T | Arg at -3 and -2 |
| PKC | R-X-S/T-X-R | Arg at -3 and +2 |
| CK2 | S/T-X-X-E/D | Acidic residue at +3 |
| MAPK | P-X-S/T-P | Pro at -2 and +1 |
| contrast | regulation | modAA | Freq |
|---|---|---|---|
| KO_vs_WT | downregulated | S | 309 |
| KO_vs_WT_at_Early | downregulated | S | 170 |
| KO_vs_WT_at_Late | downregulated | S | 221 |
| KO_vs_WT_at_Uninfect | downregulated | S | 72 |
| KO_vs_WT | upregulated | S | 499 |
| KO_vs_WT_at_Early | upregulated | S | 236 |
| KO_vs_WT_at_Late | upregulated | S | 315 |
| KO_vs_WT_at_Uninfect | upregulated | S | 140 |
| KO_vs_WT | downregulated | T | 85 |
| KO_vs_WT_at_Early | downregulated | T | 32 |
| KO_vs_WT_at_Late | downregulated | T | 79 |
| KO_vs_WT_at_Uninfect | downregulated | T | 12 |
| KO_vs_WT | upregulated | T | 97 |
| KO_vs_WT_at_Early | upregulated | T | 52 |
| KO_vs_WT_at_Late | upregulated | T | 65 |
| KO_vs_WT_at_Uninfect | upregulated | T | 14 |
| KO_vs_WT | downregulated | Y | 10 |
| KO_vs_WT_at_Early | downregulated | Y | 7 |
| KO_vs_WT_at_Late | downregulated | Y | 5 |
| KO_vs_WT_at_Uninfect | downregulated | Y | 1 |
| KO_vs_WT | upregulated | Y | 42 |
| KO_vs_WT_at_Early | upregulated | Y | 43 |
| KO_vs_WT_at_Late | upregulated | Y | 41 |
| KO_vs_WT_at_Uninfect | upregulated | Y | 1 |