Benchmarking msqrob2 across multiple datasets
Witold E. Wolski
2026-07-29
Source:vignettes/Benchmark_msqrob2.Rmd
Benchmark_msqrob2.RmdSetup
We benchmark the msqrob2 msqrobHurdle method across
three datasets. The hurdle model produces inference using
rlm from protein intensity estimates and uses
glm to obtain estimates in those cases where no
intensity-based estimates are found.
library(QFeatures)
library(msqrob2)
my_medianPolish <- function(x, verbose = FALSE, ...) {
medpol <- stats::medpolish(x, na.rm = TRUE, trace.iter = verbose, maxiter = 10)
return(medpol$overall + medpol$col)
}
msqrob2_col_map <- c(
"name" = "protein_id",
"contrast" = "contrast",
"logFC" = "log_fc",
"t" = "t_statistic",
"pval" = "p_value",
"FDR" = "p_value_adjusted",
"avgAbd" = "avg_intensity"
)
ionstar_ground_truth <- list(
id_column = "protein_id",
positive = list(label = "ECOLI", pattern = "ECOLI"),
negative = list(label = "HUMAN", pattern = "HUMAN")
)
ionstar_contrasts_L <- makeContrast(
c("dilution.e-dilution.d=0",
"dilution.d-dilution.c=0",
"dilution.c-dilution.b=0",
"dilution.b=0"),
parameterNames = c("dilution.e",
"dilution.d",
"dilution.c",
"dilution.b"))
ionstar_contrast_map <- c(
"hurdle_dilution.e - dilution.d" = "dilution_(9/7.5)_1.2",
"hurdle_dilution.d - dilution.c" = "dilution_(7.5/6)_1.25",
"hurdle_dilution.c - dilution.b" = "dilution_(6/4.5)_1.3(3)",
"hurdle_dilution.b" = "dilution_(4.5/3)_1.5")
ionstar_abd_contrast_map <- c(
"avgAbd.e.d" = "dilution_(9/7.5)_1.2",
"avgAbd.d.c" = "dilution_(7.5/6)_1.25",
"avgAbd.c.b" = "dilution_(6/4.5)_1.3(3)",
"avgAbd.b.a" = "dilution_(4.5/3)_1.5")
#' Run msqrob2 hurdle and extract merged results
#'
#' @param lfqdataPeptideNorm normalized peptide-level LFQData
#' @param lfqdataProtein protein-level LFQData (for average abundances)
#' @param formula model formula
#' @param L contrast matrix from makeContrast
#' @param contrast_map named vector mapping hurdle contrast names to standard names
#' @param abd_contrast_map named vector mapping abundance contrast names to standard names
#' @param factor_col name of the factor column in data
run_msqrob2_hurdle <- function(lfqdataPeptideNorm, lfqdataProtein,
formula, L, contrast_map, abd_contrast_map,
factor_col) {
# Create QFeatures and aggregate
se <- prolfqua::LFQDataToSummarizedExperiment(lfqdata = lfqdataPeptideNorm)
pe <- QFeatures::QFeatures(list(peptide = se), colData = colData(se))
pe <- QFeatures::aggregateFeatures(
pe, i = "peptide", fcol = "protein_Id",
name = "protein", fun = my_medianPolish)
# Fit hurdle model
prlm <- msqrobHurdle(pe, i = "protein", formula = formula, overwrite = TRUE)
prlm <- hypothesisTestHurdle(prlm, i = "protein", L, overwrite = TRUE)
# Extract hurdle results
xx <- rowData(prlm[["protein"]])
hurdle <- xx[grepl("hurdle_", names(xx))]
res <- list()
for (i in names(hurdle)) {
hurdle[[i]]$contrast <- i
res[[i]] <- prolfqua::matrix_to_tibble(hurdle[[i]], preserve_row_names = "name")
}
hurdle <- dplyr::bind_rows(res)
# Merge intensity and count models
logFC <- hurdle |> dplyr::select("name", "contrast", starts_with("logFC"))
logFC <- dplyr::filter(logFC, !is.na(logFCt))
logFC$modelName <- "msqrobHurdleIntensity"
names(logFC) <- c("name", "contrast", "logFC", "se", "df", "t", "pval", "modelName")
logOR <- hurdle |> dplyr::select("name", "contrast", starts_with("logOR"))
logOR$modelName <- "msqrobHurdleCount"
names(logOR) <- c("name", "contrast", "logFC", "se", "df", "t", "pval", "modelName")
ddd <- dplyr::anti_join(logOR, logFC, by = c("name", "contrast"))
all <- dplyr::bind_rows(ddd, logFC) |> dplyr::arrange(contrast, name)
all <- prolfqua::adjust_p_values(all, column = "pval", group_by_col = "contrast")
# Remap contrast names
all <- all |> dplyr::mutate(
contrast = dplyr::case_when(
contrast %in% names(contrast_map) ~ contrast_map[contrast],
TRUE ~ contrast))
stopifnot(sum(all$contrast == "something wrong") == 0)
# Compute average abundances per contrast
st <- lfqdataProtein$get_Stats()
protAbundanceIngroup <- st$stats()
protAbundanceIngroup <- protAbundanceIngroup |>
tidyr::pivot_wider(
id_cols = protein_Id,
names_from = !!rlang::sym(factor_col),
names_prefix = "abd.",
values_from = meanAbundance)
# Compute average abundances for each contrast pair
abd_cols <- grep("^abd\\.", names(protAbundanceIngroup), value = TRUE)
abd_values <- sort(abd_cols)
for (nm in names(abd_contrast_map)) {
# Extract the two group suffixes from the abd_contrast_map name (e.g., "avgAbd.e.d" -> "e", "d")
parts <- strsplit(sub("^avgAbd\\.", "", nm), "\\.")[[1]]
col1 <- paste0("abd.", parts[1])
col2 <- paste0("abd.", parts[2])
protAbundanceIngroup <- protAbundanceIngroup |>
dplyr::mutate(!!nm := mean(c(.data[[col1]], .data[[col2]]), na.rm = TRUE))
}
protAbundanceIngroup <- protAbundanceIngroup |>
dplyr::select(-starts_with("abd")) |>
tidyr::pivot_longer(starts_with("avgAbd"), names_to = "contrast", values_to = "avgAbd")
protAbundanceIngroup <- protAbundanceIngroup |>
dplyr::mutate(contrast = dplyr::case_when(
contrast %in% names(abd_contrast_map) ~ abd_contrast_map[contrast],
TRUE ~ contrast))
bb <- dplyr::inner_join(all, protAbundanceIngroup,
by = c("name" = "protein_Id", "contrast" = "contrast"))
bb
}Dataset 1: IonStar / MaxQuant peptides
datadir <- file.path(find.package("prolfquadata"), "quantdata")
inputMQfile <- file.path(datadir, "MAXQuant_IonStar2018_PXD003881.zip")
inputAnnotation <- file.path(datadir, "annotation_Ionstar2018_PXD003881.xlsx")
mqdata <- list()
mqdata$data <- prolfquapp::tidyMQ_Peptides(inputMQfile)
mqdata$config <- create_mq_peptide_config()
annotation <- readxl::read_xlsx(inputAnnotation)
data <- dplyr::inner_join(mqdata$data, annotation, by = "raw.file")
mqdata$config$factors[["dilution."]] <- "sample"
mqdata$config$factors[["run_Id"]] <- "run_ID"
mqdata$config$factor_depth <- 1
mqdata$data <- prolfqua::setup_analysis(data, mqdata$config)
lfqdata_mq <- prolfqua::LFQData$new(mqdata$data, mqdata$config)
lfqdata_mq$set_data(lfqdata_mq$data_long() |> dplyr::filter(!grepl("^REV__|^CON__", protein_Id)))
lfqdata_mq$filter_proteins_by_peptide_count()
lfqdata_mq$remove_small_intensities()
lfqdata_mq$hierarchy_counts()## # A tibble: 1 × 3
## isotope protein_Id peptide_Id
## <chr> <int> <int>
## 1 light 4178 29879
tr <- lfqdata_mq$get_Transformer()
subset_h <- lfqdata_mq$get_copy()
subset_h$set_data(subset_h$data_long() |> dplyr::filter(grepl("HUMAN", protein_Id)))
subset_h <- subset_h$get_Transformer()$log2()$lfq
lfqdataNorm_mq <- tr$log2()$robscale_subset(lfqsubset = subset_h)$lfq
lfqAgg_mq <- lfqdataNorm_mq$get_Aggregator()
lfqAgg_mq$aggregate()
lfqProt_mq <- lfqAgg_mq$lfq_agg
bb_mq <- run_msqrob2_hurdle(
lfqdataPeptideNorm = lfqdataNorm_mq,
lfqdataProtein = lfqProt_mq,
formula = ~dilution.,
L = ionstar_contrasts_L,
contrast_map = ionstar_contrast_map,
abd_contrast_map = ionstar_abd_contrast_map,
factor_col = "dilution.")## | | | 0%## | |======================================================================| 100%
res_mq <- write_contrast_results(
bb_mq,
path = "../inst/Benchresults/msqrob2_IonStar_MQ",
metadata = list(
dataset = "IonStar_MaxQuant",
method = "msqrob2",
method_description = "msqrob2 hurdle on MQ peptides + median polish",
input_file = "MAXQuant_IonStar2018_PXD003881.zip/peptides.txt",
software_version = paste0("msqrob2 ", packageVersion("msqrob2")),
date = as.character(Sys.Date()),
ground_truth = ionstar_ground_truth),
col_map = msqrob2_col_map)
benchmark_mq <- benchmark_from_result(res_mq)
prolfqua::table_facade(benchmark_mq$smc$summary,
caption = "Nr of proteins with 0, 1, 2, 3 missing contrasts.")| nr_missing | protein_id |
|---|---|
| 0 | 4178 |
benchmark_mq$plot_ROC(xlim = 0.2)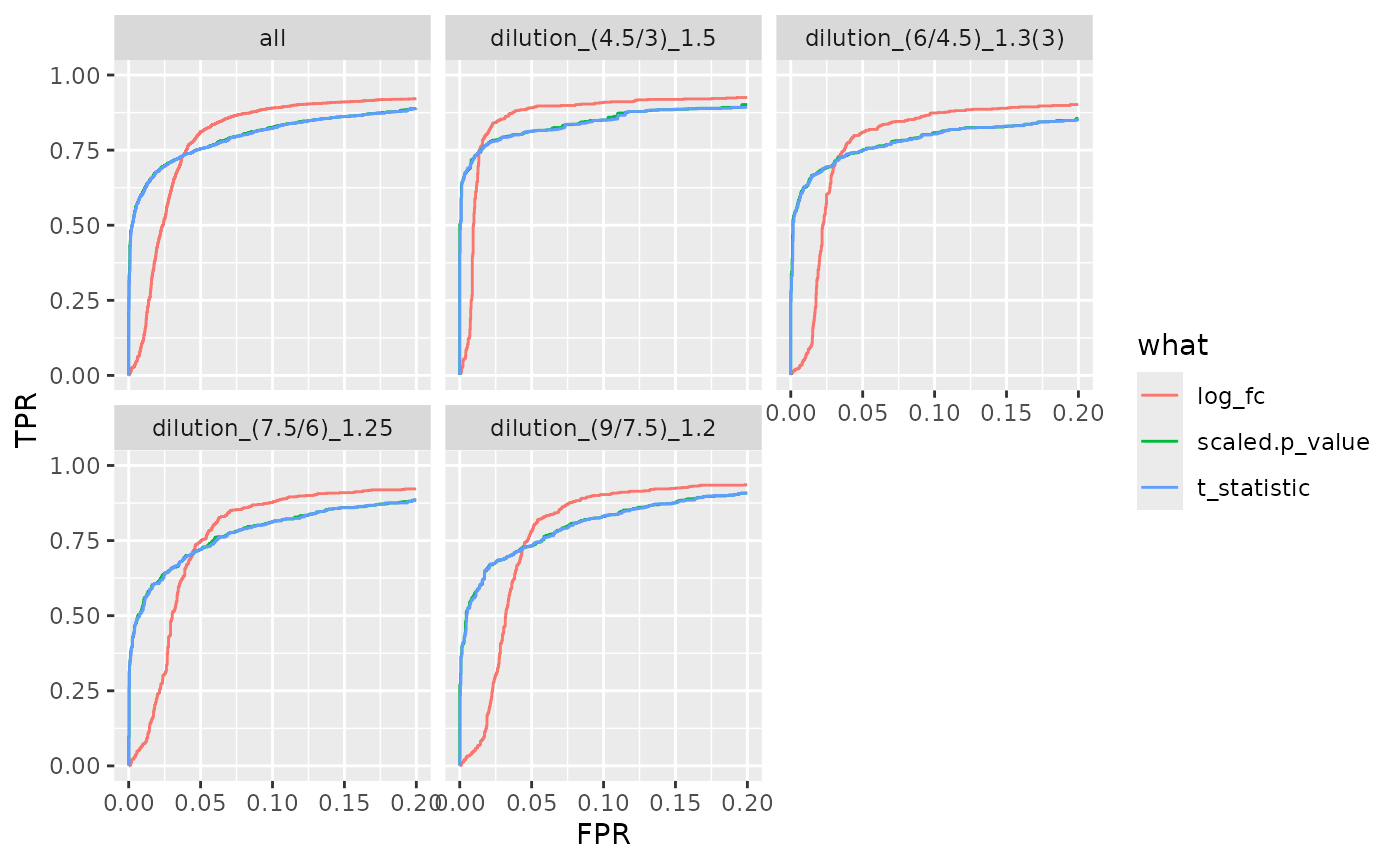
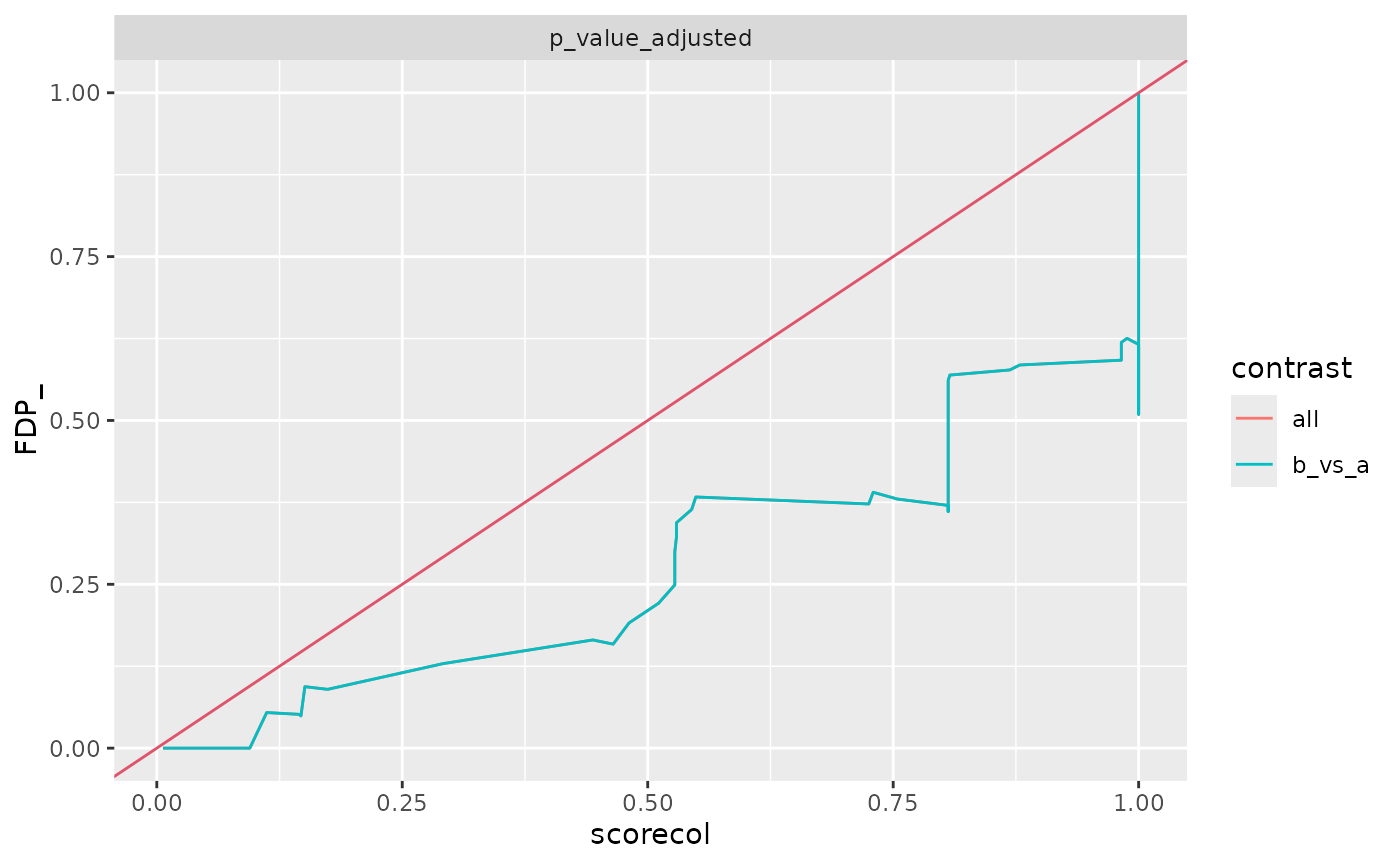
benchmark_mq$plot_FDRvsFDP()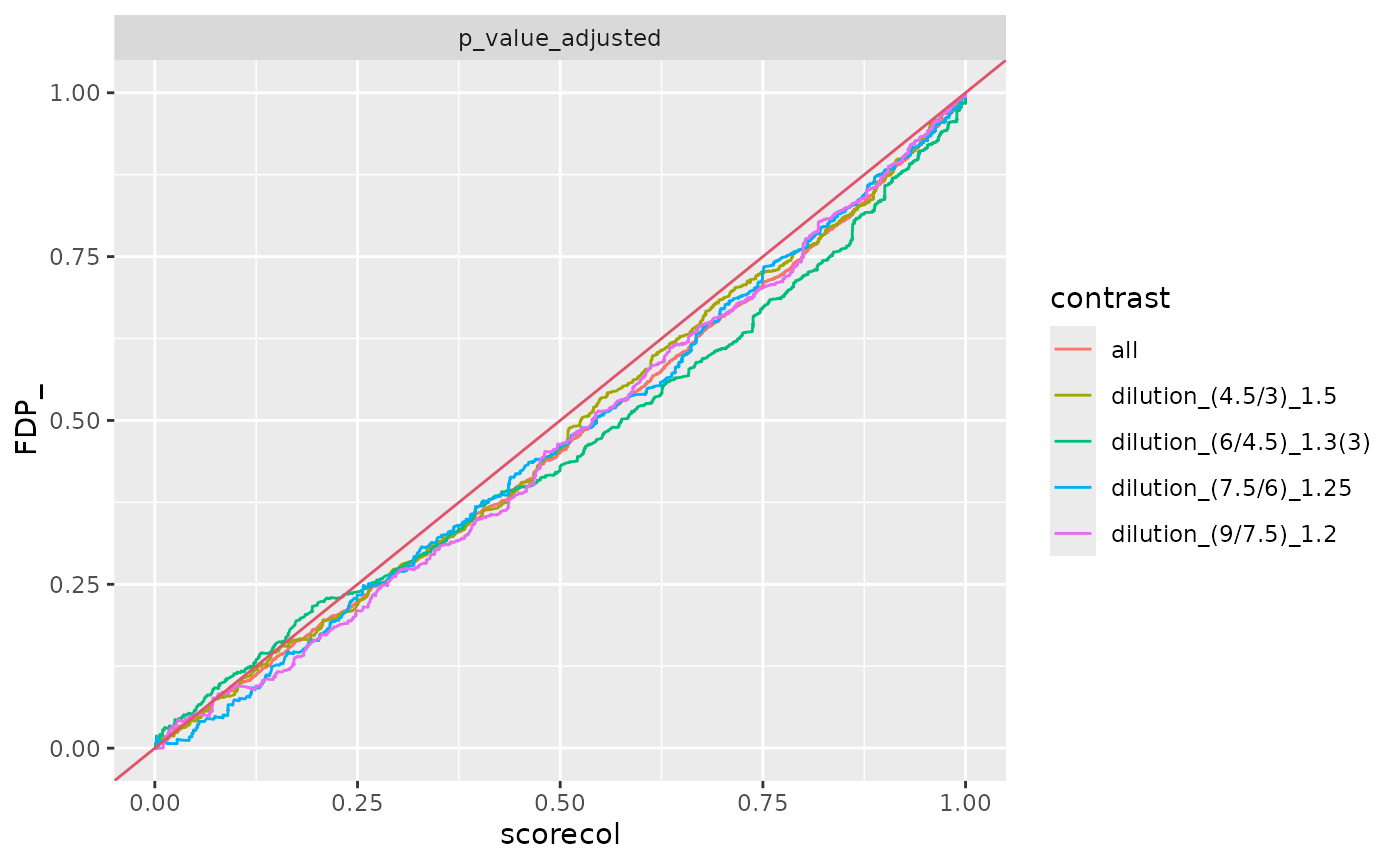
Dataset 2: IonStar / FragPipe MSstats.csv
inputFragfile <- file.path(datadir, "MSFragger_IonStar2018_PXD003881.zip")
annotation_fp <- readxl::read_xlsx(inputAnnotation)
msstats <- read.csv(
unz(inputFragfile, "IonstarWithMSFragger/MSstats.csv"),
header = TRUE, sep = ",", stringsAsFactors = FALSE)
msstats <- tibble::tibble(msstats)
msstats$Run <- tolower(msstats$Run)
merged <- dplyr::inner_join(annotation_fp, msstats, by = c("raw.file" = "Run"))
summedPep <- merged |>
dplyr::group_by(raw.file, run_ID, sample, ProteinName, PeptideSequence) |>
dplyr::summarize(nprecursor = dplyr::n(),
pepIntensity = sum(Intensity, na.rm = TRUE), .groups = "drop")
atable <- prolfqua::AnalysisConfiguration$new()
atable$file_name <- "raw.file"
atable$hierarchy[["protein_Id"]] <- c("ProteinName")
atable$hierarchy[["peptide_Id"]] <- c("PeptideSequence")
atable$hierarchy_depth <- 1
atable$set_response("pepIntensity")
atable$factors[["dilution."]] <- "sample"
atable$factors[["run"]] <- "run_ID"
atable$factor_depth <- 1
config_fp <- atable
adata_fp <- prolfqua::setup_analysis(summedPep, config_fp)
lfqdata_fp <- prolfqua::LFQData$new(adata_fp, config_fp)
lfqdata_fp$hierarchy_counts()## # A tibble: 1 × 3
## isotopeLabel protein_Id peptide_Id
## <chr> <int> <int>
## 1 light 5122 31107
lfqdata_fp$filter_proteins_by_peptide_count()
lfqdata_fp$remove_small_intensities()
knitr::kable(lfqdata_fp$hierarchy_counts(), caption = "number of proteins and peptides.")| isotopeLabel | protein_Id | peptide_Id |
|---|---|---|
| light | 3904 | 29889 |
pl <- lfqdata_fp$get_Plotter()
pl$intensity_distribution_density()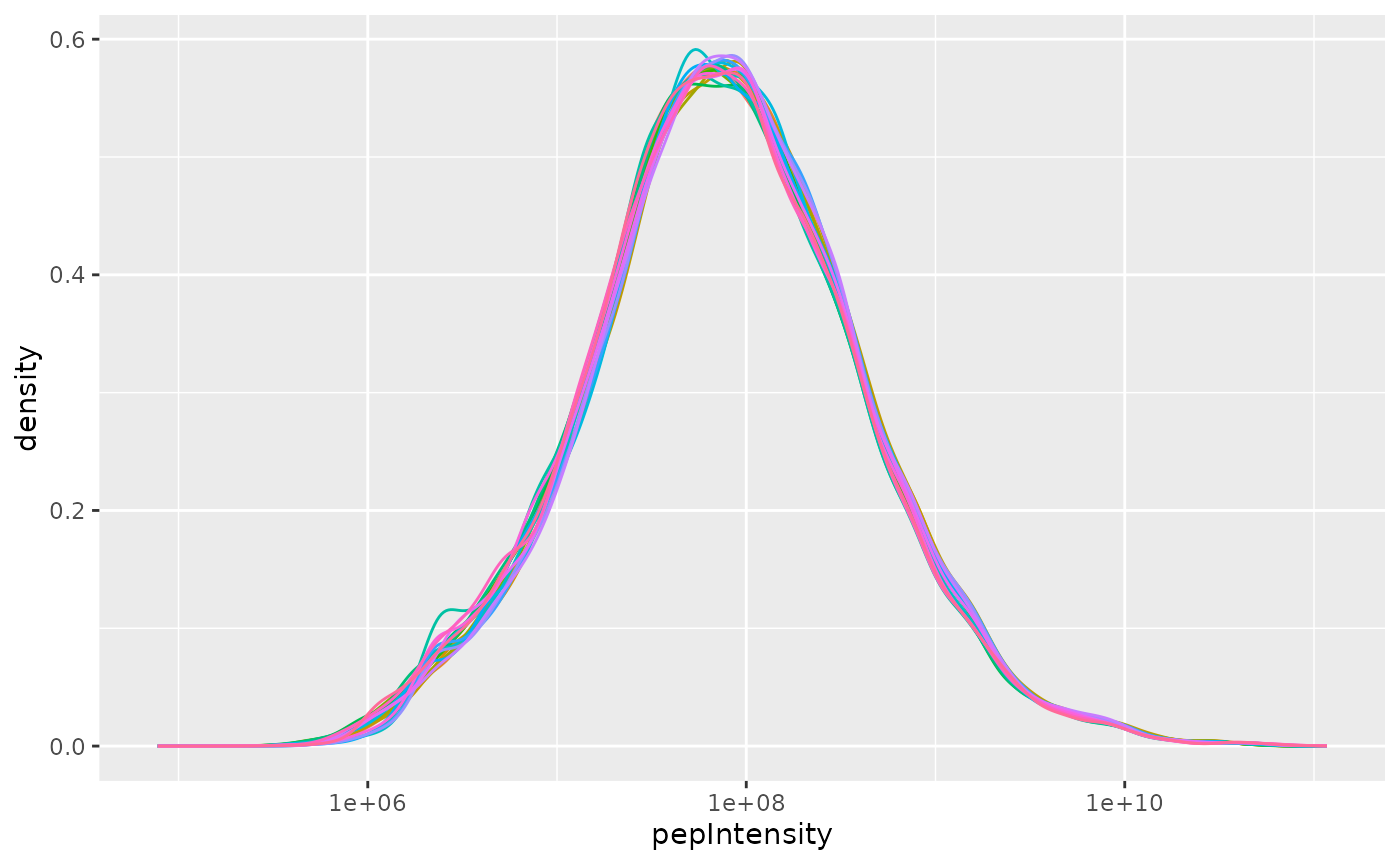
subset_h <- lfqdata_fp$get_copy()$get_Transformer()$log2()$lfq
subset_h$set_data(subset_h$data_long() |> dplyr::filter(grepl("HUMAN", protein_Id)))
tr <- lfqdata_fp$get_Transformer()
lfqdataNorm_fp <- tr$log2()$robscale_subset(lfqsubset = subset_h)$lfq
agg_fp <- lfqdataNorm_fp$get_Aggregator()
agg_fp$aggregate()
lfqProt_fp <- agg_fp$lfq_agg
lfqProt_fp$hierarchy_counts()## # A tibble: 1 × 2
## isotopeLabel protein_Id
## <chr> <int>
## 1 light 3904
bb_fp <- run_msqrob2_hurdle(
lfqdataPeptideNorm = lfqdataNorm_fp,
lfqdataProtein = lfqProt_fp,
formula = ~dilution.,
L = ionstar_contrasts_L,
contrast_map = ionstar_contrast_map,
abd_contrast_map = ionstar_abd_contrast_map,
factor_col = "dilution.")## | | | 0%## | |======================================================================| 100%
res_fp <- write_contrast_results(
bb_fp,
path = "../inst/Benchresults/msqrob2_IonStar_FragPipe",
metadata = list(
dataset = "IonStar_FragPipe",
method = "msqrob2",
method_description = "msqrob2 hurdle on FragPipe MSstats.csv peptides + median polish",
input_file = "MSFragger_IonStar2018_PXD003881.zip/IonstarWithMSFragger/MSstats.csv",
software_version = paste0("msqrob2 ", packageVersion("msqrob2")),
date = as.character(Sys.Date()),
ground_truth = ionstar_ground_truth),
col_map = msqrob2_col_map)
benchmark_fp <- benchmark_from_result(res_fp)
prolfqua::table_facade(benchmark_fp$smc$summary,
caption = "Nr of proteins with 0, 1, 2, 3 missing contrasts.")| nr_missing | protein_id |
|---|---|
| 0 | 3899 |
benchmark_fp$plot_ROC(xlim = 0.2)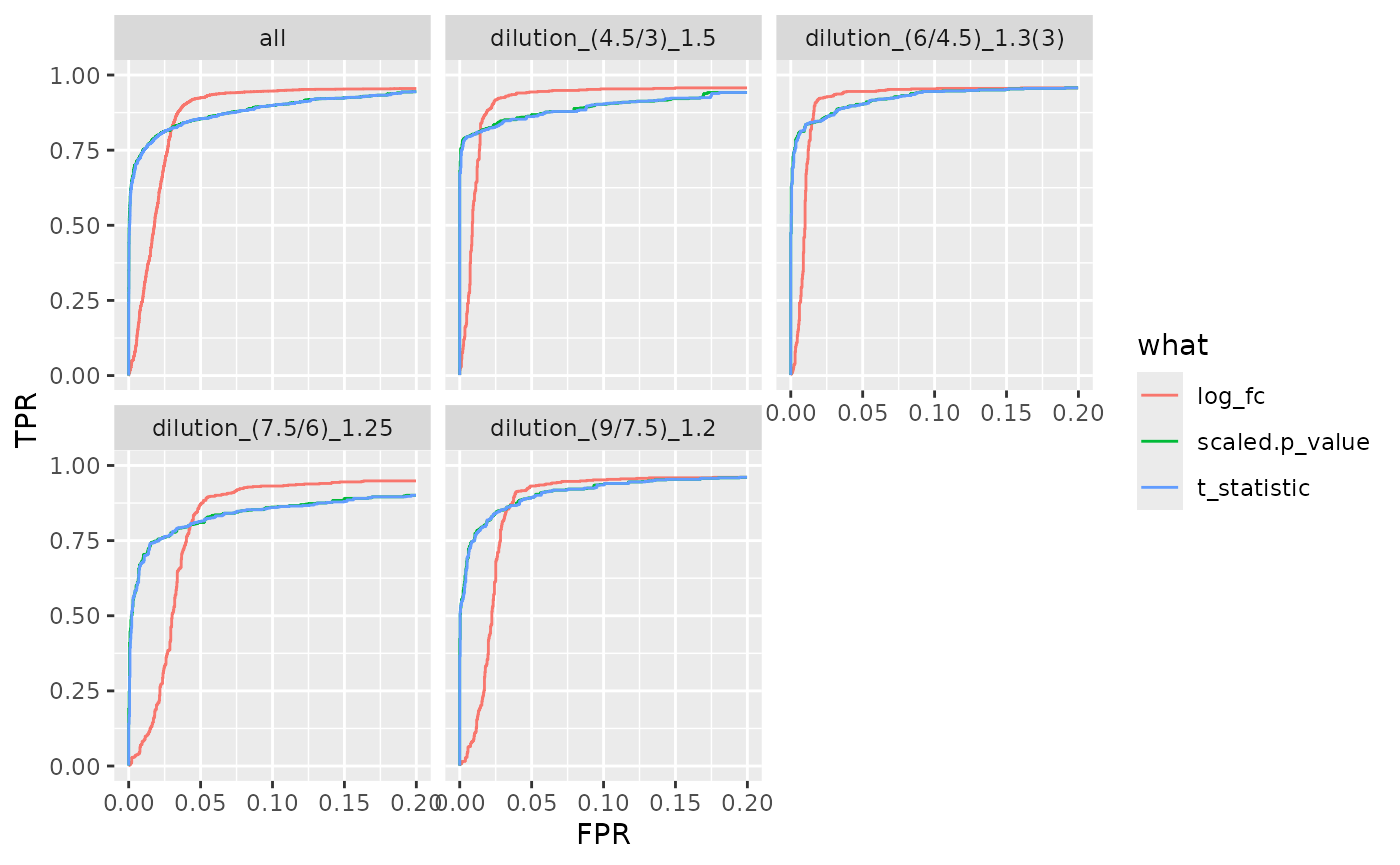
benchmark_fp$plot_FDRvsFDP()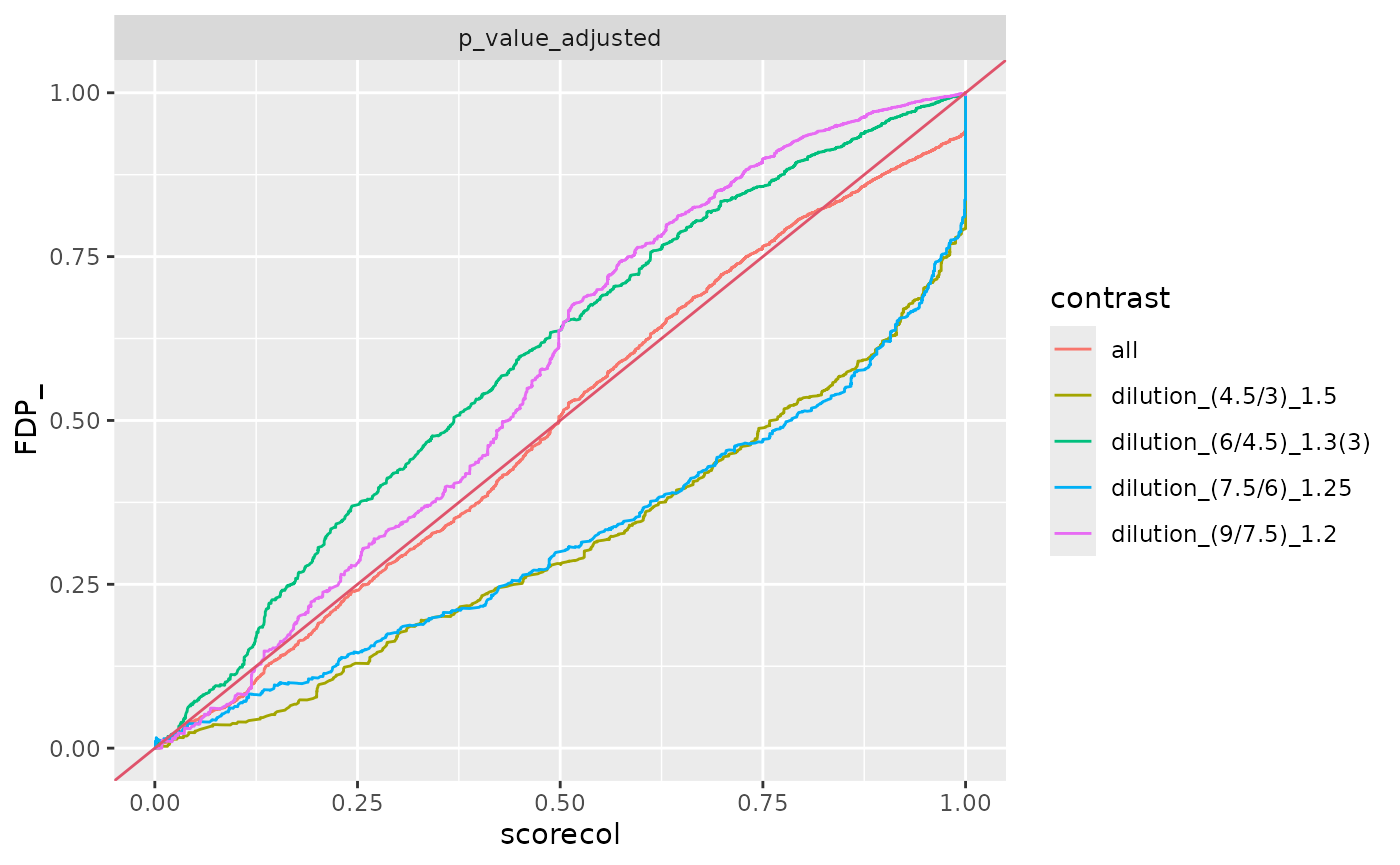
Dataset 3: CPTAC
evalCPTAC <- requireNamespace("msdata", quietly = TRUE) && evalAll
tmp <- msdata::quant(full.names = TRUE)
xx <- read.csv(tmp, sep = "\t")
peptides <- prolfquapp::tidyMQ_Peptides(xx)
annotation_cptac <- data.frame(raw.file = unique(peptides$raw.file))
annotation_cptac <- annotation_cptac |>
dplyr::mutate(group = gsub("^6", "", raw.file)) |>
dplyr::mutate(group = gsub("_[0-9]$", "", group))
peptides <- dplyr::inner_join(annotation_cptac, peptides)
config_cptac <- create_mq_peptide_config()
config_cptac$factors["group."] <- "group"
peptides <- prolfqua::setup_analysis(peptides, config_cptac)
lfqpeptides <- prolfqua::LFQData$new(peptides, config_cptac)
lfqpeptides$filter_proteins_by_peptide_count()
lfqpeptides$remove_small_intensities()
lfqpeptides$hierarchy_counts()## # A tibble: 1 × 3
## isotope protein_Id peptide_Id
## <chr> <int> <int>
## 1 light 1341 7787
tr <- lfqpeptides$get_Transformer()
tr <- tr$intensity_matrix(vsn::justvsn)
lfqtransformed <- tr$lfq
agg <- lfqtransformed$get_Aggregator()
agg$aggregate()
lfqProt_cptac <- agg$lfq_agg
# CPTAC has a single contrast: group.b vs group.a
L_cptac <- makeContrast(c("group.b=0"), parameterNames = c("group.b"))
cptac_contrast_map <- c("hurdle_group.b" = "b_vs_a")
cptac_abd_contrast_map <- c("avgAbd.b.a" = "b_vs_a")
se <- prolfqua::LFQDataToSummarizedExperiment(lfqtransformed)
pe <- QFeatures::QFeatures(list(peptide = se), colData = colData(se))
pe <- QFeatures::aggregateFeatures(
pe, i = "peptide", fcol = "protein_Id",
name = "protein", fun = my_medianPolish)## | | | 0%## | |======================================================================| 100%
prlm <- msqrobHurdle(pe, i = "protein", formula = ~group., overwrite = TRUE)
prlm <- hypothesisTestHurdle(prlm, i = "protein", L_cptac, overwrite = TRUE)
xx <- rowData(prlm[["protein"]])
hurdle <- xx[grepl("hurdle_", names(xx))]
res <- list()
for (i in names(hurdle)) {
hurdle[[i]]$contrast <- i
res[[i]] <- prolfqua::matrix_to_tibble(hurdle[[i]], preserve_row_names = "name")
}
hurdle <- dplyr::bind_rows(res)
logFC <- hurdle |> dplyr::select("name", "contrast", starts_with("logFC"))
logFC <- dplyr::filter(logFC, !is.na(logFCt))
logFC$modelName <- "msqrobHurdleIntensity"
names(logFC) <- c("name", "contrast", "logFC", "se", "df", "t", "pval", "modelName")
logOR <- hurdle |> dplyr::select("name", "contrast", starts_with("logOR"))
logOR$modelName <- "msqrobHurdleCount"
names(logOR) <- c("name", "contrast", "logFC", "se", "df", "t", "pval", "modelName")
ddd <- dplyr::anti_join(logOR, logFC, by = c("name", "contrast"))
all <- dplyr::bind_rows(ddd, logFC) |> dplyr::arrange(contrast, name)
all <- prolfqua::adjust_p_values(all, column = "pval", group_by_col = "contrast")
all$contrast <- "b_vs_a"
st <- lfqProt_cptac$get_Stats()
protAbundanceIngroup <- st$stats()
protAbundanceIngroup <- protAbundanceIngroup |>
tidyr::pivot_wider(id_cols = protein_Id,
names_from = group., names_prefix = "abd.",
values_from = meanAbundance)
protAbundanceIngroup <- protAbundanceIngroup |>
dplyr::mutate(avgAbd.b.a = mean(c(abd.b, abd.a), na.rm = TRUE))
protAbundanceIngroup <- protAbundanceIngroup |>
dplyr::select(-starts_with("abd")) |>
tidyr::pivot_longer(starts_with("avgAbd"), names_to = "contrast", values_to = "avgAbd")
protAbundanceIngroup$contrast <- "b_vs_a"
bb_cptac <- dplyr::inner_join(all, protAbundanceIngroup,
by = c("name" = "protein_Id", "contrast" = "contrast"))
res_cptac <- write_contrast_results(
bb_cptac,
path = "../inst/Benchresults/msqrob2_CPTAC",
metadata = list(
dataset = "CPTAC",
method = "msqrob2",
method_description = "msqrob2 hurdle on CPTAC MQ peptides + VSN + median polish",
input_file = "msdata::quant()",
software_version = paste0("msqrob2 ", packageVersion("msqrob2")),
date = as.character(Sys.Date()),
ground_truth = list(
id_column = "protein_id",
positive = list(label = "UPS", pattern = "_UPS"),
negative = list(label = "YEAST", pattern = "_YEAST")
)),
col_map = msqrob2_col_map)
benchmark_cptac <- benchmark_from_result(res_cptac)
prolfqua::table_facade(benchmark_cptac$smc$summary,
caption = "Nr of proteins with 0, 1, 2, 3 missing contrasts.")| nr_missing | protein_id |
|---|---|
| 0 | 1333 |
benchmark_cptac$plot_ROC(xlim = 0.2)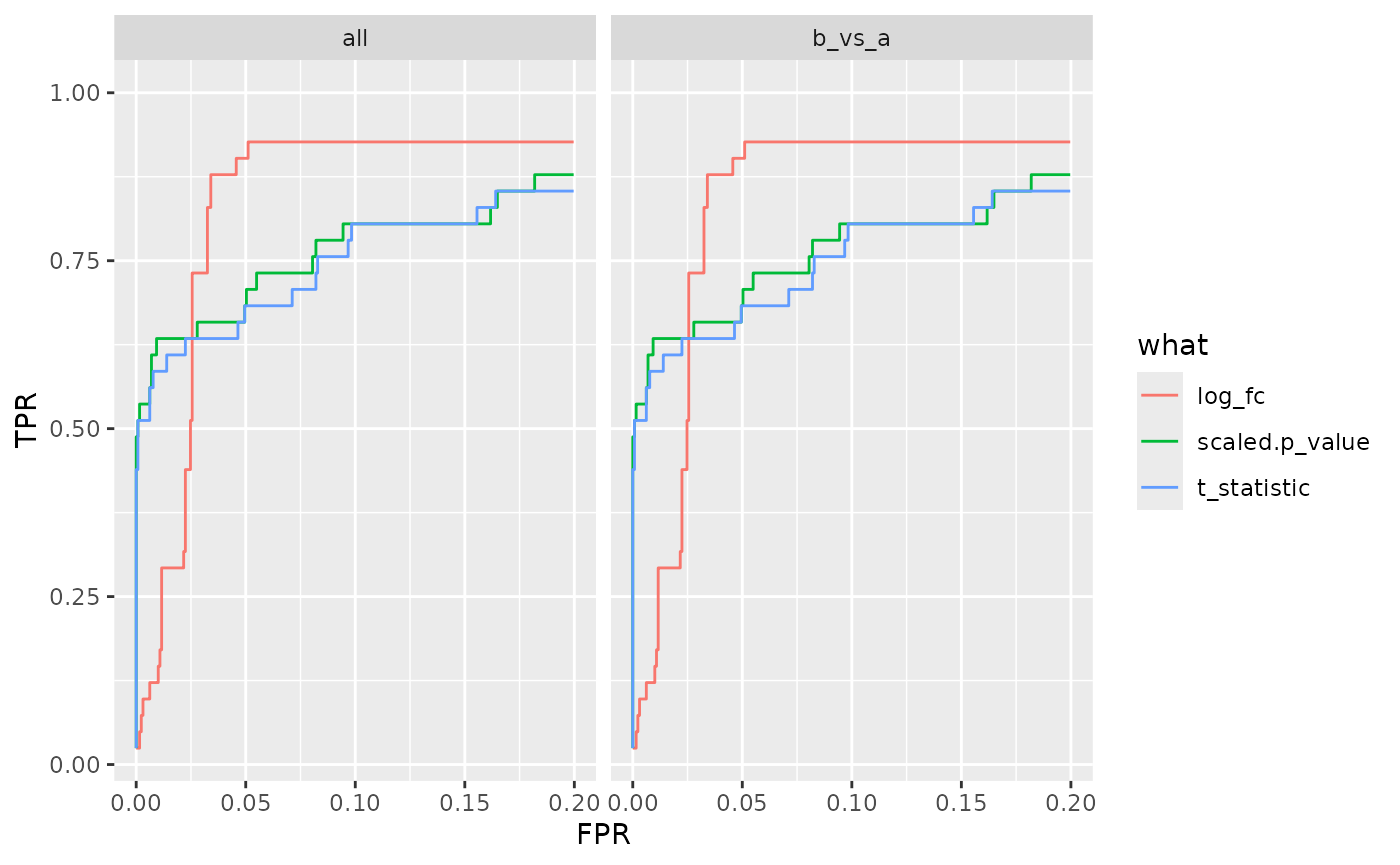
benchmark_cptac$plot_FDRvsFDP()